It is not uncommon to see more than one morphological presentation in the same patient. With disease progression, patches may evolve into plaques. In this phase (mycotic), the lesions become infiltrated and present as well-demarcated, red or reddish-brown to purple plaques. Central clearing may develop, resulting in unusual configurations (e.g. serpiginous, arciform, horseshoe). The tumour stage is characterized by raised nodules or tumours; these are flesh-coloured or brown to bluish-red lesions covered with a thin, stretched, atrophic-appearing epidermis, with or without telangiectasia. Lesions often undergo necrosis or ulceration and at times may disappear spontaneously.
Lesions of MF may appear anywhere, but there is a predilection for non-sun exposed (or so-called ‘double-covered’) areas: the trunk below the waistline, buttocks, flanks, breasts, inner thighs, inner arms and periaxillary areas. Rarely, CTCL may also present with lesions on the scalp, palms and soles [17].
There are certain clinical features that differentiate paediatric from adult CTCL. In children, studies have consistently shown presentation in early-stage disease. In one study of 10 patients, all presented at stage IIB or less, including two patients with unilesional disease [18]. An interesting feature of paediatric CTCL is an over-representation of hypopigmented and poikilodermic disease (Fig. 102.1d) [3,5,12]. The hypopigmented lesions present as non-atrophic white patches, with minimal scaling [19,20]. Not uncommonly, the affected area is devoid of hair (personal observation). The incidence is as high as 70% in one paediatric study, with 43% presenting in light-skinned individuals [18]. Recent results from an international registry of paediatric CTCL patients confirmed the over-representation of early-stage disease and hypopigmented CTCL in these patients [21].
Differential Diagnosis.
Other papulosquamous (psoriasis, tinea corporis, pityriasis lichenoides chronica, lichen aureus, parapsoriasis), dermatitic (atopic dermatitis) and hypopigmentary (postinflammatory hypopigmentation and vitiligo [22]) diseases can appear morphologically similar to MF lesions.
Histopathology.
Mycosis fungoides in children is histologically and immunophenotypically indistinguishable from that seen in adults [23].
The patch stage is characterized by a superficial band-like or lichenoid infiltrate. There is epidermotropism, defined as the presence of linear or single dispersed atypical lymphocytes in the epidermis [24]. The atypical lymphocytes manifest hyperchromatic nuclei with cerebriform nuclear contours. The intraepidermal cells are typically larger and more atypical compared to their dermal counterparts. The epidermis may be variably hyperplastic but typically, spongiosis is minimal and keratinocyte necrosis is not prominent. Distinctive halo-like lacunar spaces may be seen to surround the intraepidermal cells [25], a discriminating diagnostic feature of MF (Fig. 102.2a). A variable admixture of plasma cells, eosinophils and histiocytes may be present, but these are not prominent. The dermis may show oedema or fibrosis. In the early stages of MF, architectural abnormalities are often more helpful in establishing the diagnosis than cytological features, which may not be highly atypical [26–28]. Early lesions have protean histological features, and it is not unusual for multiple biopsies to be required for diagnosis in some cases [27]. The International Society for Cutaneous Lymphoma (ISCL) has proposed a diagnostic algorithm for early MF [17].
Fig. 102.2 (a) Mycosis fungoides, patch stage. A relatively sparse dermal infiltrate of atypical lymphocytes is present. Atypical lymphocytes surrounded by clear halos are present in the basal aspect of the epidermis. Note the absence of epidermal spongiosis (H&E). (b) Mycosis fungoides. A collection of atypical lymphocytes within the epidermis, forming a Pautrier microabscess (H&E).
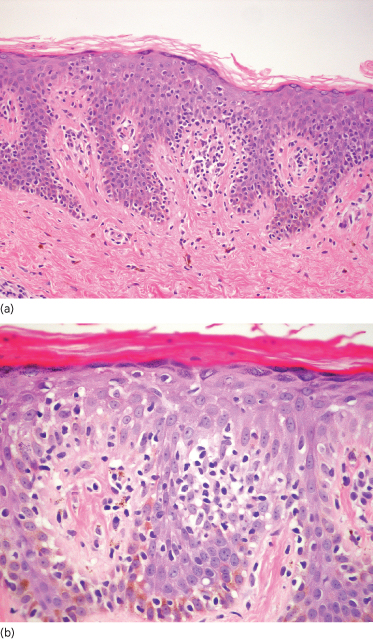
The plaque stage features a denser infiltrate with more easily identified atypical lymphocytes. There is more prominent epidermotropism. Pautrier microabscesses (sharply marginated clusters of lymphocytes within the epidermis) may be present (Fig. 102.2b).
The tumour stage shows diffuse dermal infiltrates and loss of epidermotropism and Pautrier microabscesses. The infiltrate is monomorphous and dominated by large atypical cells [29]. Mitoses are generally numerous. The infiltrate may extend into the subcutis. Large cell transformation, defined as a biopsy specimen showing large cells at least four times the size of a small lymphocyte in 25% or more of the dermal infiltrate, may occur [30–32]. This may be either CD30 negative or positive and is associated with a poor prognosis [33].
Immunophenotype.
The neoplastic cells in MF have a mature T-cell phenotype, usually of helper T-cells, and the most common immunohistochemical profile is CD2+, CD3+, CD4+, CD5+ and CD45RO+. During progression of the disease, loss of CD2, CD5 and CD7 may be seen, particularly in the epidermotropic cerebriform cells. Other immunophenotypical profiles have been described, however, without prognostic significance:
- cases with a CD4- CD8+ mature T-cell phenotype [34–37], over-represented in childhood [18,36] and in hypopigmented variants [3,38]
- cases with a CD4- CD8- phenotype [39] (6/18 cases were in the paediatric age group)
- cases with a CD56+ phenotype [40–42].
Genetic Features.
Genetic features in paediatric MF do not appear different from those in adult cases. Clonal T-cell rearrangements are not as common as in adult cases, possibly due to the patch stage presentation with relatively low infiltrate load and difficulties in doing large biopsies. Detection of a monoclonal population in early lesions of MF does not have any prognostic significance [43].
Prognosis and Predictive Factors.
Although the earlier literature suggested that paediatric MF has a more aggressive disease course and association with other forms of lymphomas [7], more recent and larger case series indicate that the natural history is stage dependent [3,4,8,12,18,19]. When matched for stage, the prognosis is similar to that in adults, although the onset of lesions of MF in childhood may increase the probability of disease progression during the lifetime of the patient [5].
Treatment.
The ideal treatment for paediatric CTCL is still unclear. To date, there are no controlled trials delineating a survival benefit from any of the therapies currently used [44]. It is always important to balance achieving disease remission and preventing long-term damage from the therapy used, particularly in a growing child. Accepted methods of treating patch and plaque stage include observation only (for isolated/asymptomatic patches), topical steroids, topical mechlorethamine, topical carmustine (BCMU) and topical bexarotene, PUVA (psoralen chemotherapy with ultraviolet light radiation), UVA1, UVB1, localized electron beam therapy and topical nitrogen mustard [45–51].
In a consensus statement from Europe, emollients with or without moderate topical steroids for localized disease were an acceptable therapeutic choice, given that life expectancy is not adversely affected [3].
The usefulness of topical options such as mechlorethamine hydrochloride 0.01% or 0.02%, carmustine (BCNU), imiquimod, tazarotene and a novel retinoid, 1% bexarotene, in the paediatric setting is limited due to the associated high rate of severe irritant dermatitis. In the few published paediatric series, the most common treatment modality was topical steroids, irrespective of the diagnostic category or staging, followed by light therapy (PUVA, UVB, narrow-band UVB) and combination therapy (light therapy in addition to topical corticosteroids) [8,12,18].
Mycosis Fungoides Variants and Subtypes
Folliculotropic Mycosis Fungoides
Folliculotropic MF (MF-associated follicular mucinosis [52]) is a variant of MF characterized by the presence of follicular/perifollicular lymphoid infiltrates. Most cases show mucinous degeneration of the hair follicles (follicular mucinosis) which is, by itself, of no clinical significance. However, the deep, follicular and perifollicular localization of the neoplastic infiltrates renders the disease more refractory to skin-targeted therapies [53]. Although folliculotropic MF occurs mostly in adults, children and adolescents may also be affected [53–55]. Clinical presentation is grouped follicular papules, acneiform lesions, indurated plaques or tumours. Lesions occur most frequently in the head and neck region [53,56]. Infiltrative plaques in the eyebrows with alopecia are characteristic. Pruritus is severe, and secondary bacterial infection is frequent.
Histology in the paediatric age group is no different from that in adults, showing perivascular and periappendageal infiltrates of small to medium-sized, sometimes large, atypical lymphocytes with hyperchromatic and cerebriform nuclei [53]. Alcian blue staining shows mucinous degeneration of the follicular epithelium in cases associated with follicular mucinosis. A mixture of eosinophils and sometimes plasma cells is often present [53]. The neoplastic cells have an immunophenotype of CD3 and CD4 positivity and CD8 negativity, similar to that seen in usual MF [56]. Small clusters of CD30+ blast cells and admixed B-cells can be seen [53]. The 5-year disease-specific survival is approximately 70–80% which is similar to that of classic tumour stage MF [53,57].
Pagetoid Reticulosis.
Pagetoid reticulosis (PR) is a distinct variant of MF characterized clinically by localized patches or plaques with an indolent behaviour, and histologically by an exclusively intraepidermal infiltrate of neoplastic T-cells. Pagetoid reticulosis is seen in both paediatric [58–61] and adult populations with a male-to-female ratio of 2 : 1 [60]. Clinically, patients present with a large, solitary, erythematous, scaly or verrucous patch or plaque, usually on the distal part of a limb. The disease is slowly progressive and extracutaneous dissemination is not seen.
Histologically, the epidermis is irregularly acanthotic with overlying hyperkeratosis and spotty parakeratosis [60]. There is epidermal infiltration by singly dispersed or clustered medium-sized to large atypical lymphoid cells with pale eosinophilic or vacuolated cytoplasm and hyperchromatic and cerebriform nuclei [60]. Accompanying Langerhans cells and histiocytes are also present [62]. The neoplastic epidermal lymphoid infiltrate is composed of T-cells which may have a CD4+ T-helper phenotype, CD8+ T-cytotoxic/suppressor phenotype or a CD4/CD8 double-negative phenotype [63–65].
Granulomatous Slack Skin.
Granulomatous slack skin (GSS) is a very rare subtype of CTCL. It is a clinicopathological entity characterized by development of areas of pendulous, lax skin in the major skinfolds (especially axillae and groins) [66]. Males are predominantly affected [67]. While the majority of cases have been reported in adults, cases in adolescents/younger adults aged 14–20 years have also been seen [68–72], with similar clinical behaviour and histological appearance.
Histology reveals a dense granulomatous dermal infiltrate containing atypical T-cells with variably cerebriform nuclei, macrophages and numerous multinucleated giant cells containing a large number of nuclei [67]. The atypical T-cells have a CD3+, CD4+ and CD8- immunophenotype [71]. The multinucleated giant cells are reactive for lysozyme, CD68 and S-100 [72]. Destruction of elastic fibres is characteristic and accounts for the formation of the pendulous skinfolds.
Most cases have an indolent course with long survival. However, there is an association with Hodgkin disease in one-third of patients [66,68,73]. The pendulous skinfolds tend to recur after surgical excision [74].
Sézary Syndrome
Sézary syndrome (SS) is a distinctive erythrodermic cutaneous T-cell lymphoma characterized by pruritic erythroderma, generalized lymphadenopathy and circulating malignant T-cells in the peripheral blood. It is a rare condition that occurs almost exclusively in adults. Meister et al. described an 11-year-old girl who had generalized exfoliative erythroderma, intense pruritus, peripheral lymphadenopathy, mycosis cells in the skin and lymph nodes, and Sézary cells in the peripheral blood [75]. LeBoit et al. report a 12-year-old girl who had erythrodermic follicular mucinosis, hypereosinophilia, circulating Sézary cells, and both immunophenotypical and genotypical evidence of T-cell neoplasia, commenting that erythrodermic follicular mucinosis may represent an unusual variant of the Sézary syndrome [76].
Adult T-Cell Leukaemia/Lymphoma
Adult T-cell leukaemia/lymphoma is a T-cell neoplasm aetiologically associated with the type C retrovirus human T-cell leukaemia virus 1 (HTLV-1). Although the condition is seen predominantly in adults [77–81], cases occurring in children have been reported [82,83]. It is endemic in areas with a high prevalence of HTLV-1 in the population, such as Japan, the Caribbean Islands, South America and parts of Central Africa. It develops in 1–5% of seropositive individuals after more than two decades of viral persistence. Skin lesions may resemble those of MF clinically (papules, plaques and tumours) [78] and histologically. There is a superficial band-like infiltrate of medium-sized to large T lymphoid cells with prominent epidermotropism and intraepidermal Pautrier-like microabscesses. In addition, epithelial necrosis and angiocentricity with angio-invasion are often seen [78,80]. Skin lesions in the smouldering variant may contain only sparse neoplastic cells with mild atypia. The neoplastic T-cells have a CD3+, CD4+, CD8- phenotype with expression of CD25 [77,84].
Primary Cutaneous CD30+ Lymphoproliferative Disorders
Primary cutaneous CD30+ lymphoproliferative disorders are the second most common group of cutaneous T-cell lymphomas [1]. However, in children under 20 years of age, equal numbers of MF and primary cutaneous CD30+ lymphoproliferative disorders were noted [5]. Primary cutaneous CD30+ lymphoproliferative disorders comprise a clinical and morphological spectrum of diseases which include lymphomatoid papulosis (LyP) and primary cutaneous anaplastic large cell lymphoma (C-ALCL) at two ends of the spectrum. Despite good clinicopathological correlation, some cases cannot be easily classified as one of the two [85]. Other disease entities falling into this spectrum include Hodgkin disease, MF with large cell transformation, CD30+ large B-cell lymphoma, and CD30+ lymphomatoid drug reactions [86]. The feature common to all is the expression of CD30 which is a cytokine receptor belonging to the tumour necrosis factor receptor superfamily [87].
Primary Cutaneous Anaplastic Large Cell Lymphoma
Definition.
Primary cutaneous anaplastic large cell lymphoma (C-ALCL) is a lymphoid neoplasm composed of large cells with an anaplastic, pleomorphic or immunoblastic cytomorphology with expression of CD30 by the majority (greater than 75%) of the tumour cells [85]. There should be no evidence of prior or concurrent lymphomatoid papulosis, MF or any other form of cutaneous lymphoma [85]. Cutaneous ALCL is different clinically and biologically from systemic anaplastic large cell lymphoma (S-ALCL), from which it must be distinguished [88,89].
Epidemiology.
Cutaneous ALCL generally occurs in adults, and is rare in the paediatric age group [88,90–96]. In contrast, S-ALCL is a relatively common lymphoma in this age group, and has a propensity for cutaneous involvement. C-ALCL may be seen in HIV-infected individuals [88].
Clinical Features.
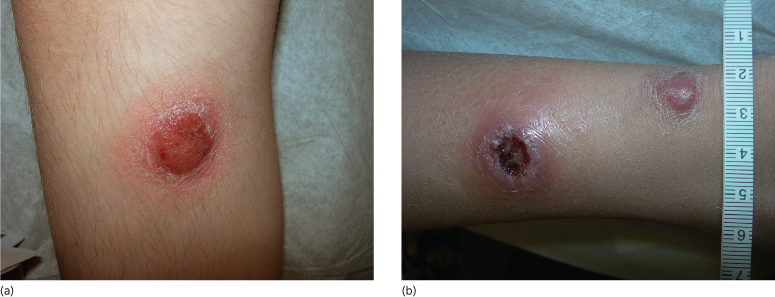
Patients have a history of rapidly growing asymptomatic, solitary or multiple skin nodules/tumours with a violaceous colour. Most lesions have a tendency to ulcerate (Fig. 102.3) [92].
Fig. 102.3 (a) Primary cutaneous anaplastic large cell lymphoma. Large tumour, with superficial ulceration. (b) Primary cutaneous anaplastic large cell lymphoma. Two large violaceous tumours, one intact, one with a deep central ulceration.
Histopathology.
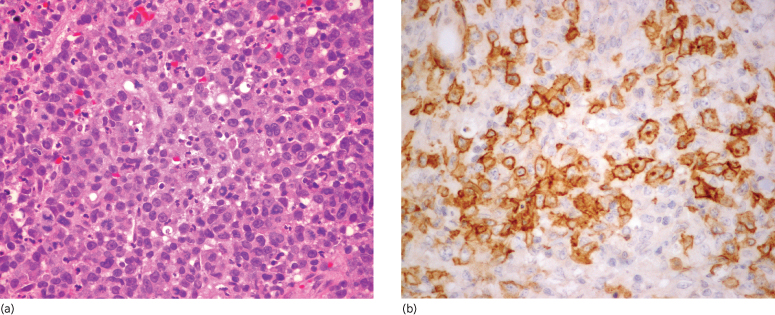
The tumour consists of a diffuse, cohesive and sheet-like infiltrate of large lymphoid cells which are CD30+. The infiltrate is dense and usually extends through all levels of the dermis and often into the subcutis. In most cases, the tumour cells appear anaplastic, displaying eccentric, round/oval or irregularly shaped nuclei and prominent nucleoli (Fig. 102.4). The cytoplasm is abundant and eosinophilic, and prominent inclusion-like Golgi accentuation may be seen [88]. Erythrophagocytosis by tumour cells has been reported [88,89]. The background contains a scant inflammatory cell infiltrate. Less commonly, the tumour cells have a non-anaplastic (pleomorphic or immunoblastic) appearance [90,97].
Fig. 102.4 (a) Primary cutaneous anaplastic large cell lymphoma. Cohesive sheets of large, anaplastic cells with admixed inflammatory cells (H&E). (b) Primary cutaneous anaplastic large cell lymphoma. CD30 is expressed by the majority of tumour cells. In addition to membranous staining, some cells display a dot-like paranuclear Golgi staining pattern (immunoperoxidase stain for CD30).
Immunophenotype.
By definition, CD30 must be expressed by at least 75% of the large lymphoid tumour cells. The tumour cells usually show an activated CD4+ T-cell phenotype with frequent expression of cytotoxic proteins (granzyme B, TIA-1 and perforin) [98,99]. There may be variable loss of CD2, CD3 or CD5 expression [89]. Unlike S-ALCL, most C-ALCLs do not express epithelial membrane antigen (EMA) [89] or anaplastic lymphoma kinase (ALK-1) [100]. Unlike Reed–Sternberg cells of classic Hodgkin disease, staining for CD15 is usually negative [95].
Genetic Features.
The t(2;5)(p23;q35) translocation, characteristically seen in S-ALCL, is not usually found in C-ALCL [101] although a case report to the contrary exists [94].
Prognosis and Predictive Factors.
The prognosis is very favourable, with an estimated survival of >90% at 5 years. A large number of paediatric cases resolve spontaneously without any intervention [102]. Skin manifestation only, without extracutaneous spread, is considered to be associated with better prognosis.
Treatment.
When intervention is warranted, topical or intralesional corticosteroids, local excision and/or radiotherapy may be employed [90].
Lymphomatoid Papulosis
Definition.
Lymphomatoid papulosis (LyP) was first described by Macaulay [103] in 1968 as an enigmatic recurring self-healing eruption which was clinically benign but histologically malignant. It is a chronic recurrent lymphoproliferative skin disease with a generally indolent clinical course, characterized by self-regressing papulonodular skin lesions with histological features of CD30+ lymphocytic cells with a variable admixed inflammatory background.
Epidemiology.
The prevalence of LyP is estimated at 1.2–1.9 cases per 1,000,000 people [104]. Although uncommon, the reported cases of childhood LyP do not represent the true incidence of the disease. Boys are reported to have an earlier onset of LyP compared to girls [105].
Clinical Features.
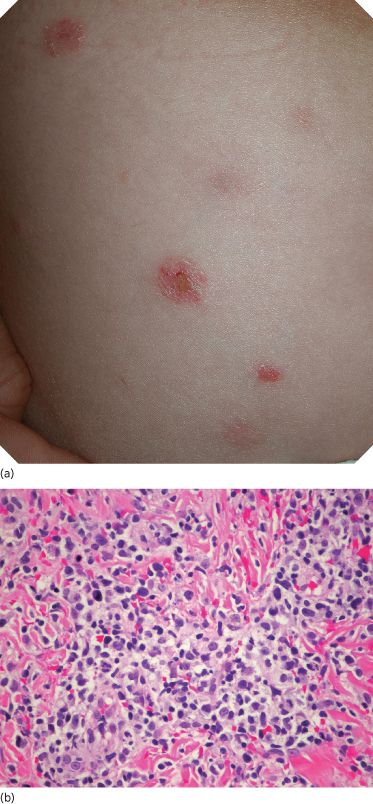
Lymphomatoid papulosis is characterized by recurrent crops of lesions that last for 3–8 weeks. The number of lesions per crop varies from patient to patient. In some, the number decreases after the initial crop. Morphologically, the LyP lesions consist of a red papule/nodule that becomes haemorrhagic/necrotic (Fig. 102.5a) a few days after the eruption and resolves with a varioliform scar on a dyspigmented background [105]. The common sites of involvement are the trunk and extremities [106], although other locations have also been documented [107]. The literature suggests that cases of regional LyP featuring clusters of lesions localized to one anatomical region are more often found in the paediatric age group than in adults [107–109].
Fig. 102.5 (a) Lymphomatoid papulosis. Multiple, scattered papules/nodules, with tendency for ulceration and scarring. (b) Lymphomatoid papulosis, type A. Scattered or small clusters of large cells are intermingled with numerous mixed inflammatory cells (H&E).
Histopathology.
The histological features of LyP are variable, and depend in part on the age of the biopsied cutaneous lesions. Three histological subtypes (A, B and C) are described, and these represent a spectrum of diseases with overlapping features.
LyP type A (histiocytic type) is the most common histological manifestation, accounting for approximately 75% of LyP cases [90,108]. A similar proportion was seen in a series of paediatric patients [105]. In fully developed cases, a wedge-shaped dermal infiltrate is seen. This consists of medium-sized to large pleomorphic or anaplastic lymphoid cells which may be scattered or grouped in small clusters (Fig. 102.5b). Mitoses are usually numerous. Numerous admixed inflammatory cells (neutrophils, eosinophils, lymphocytes and histiocytes) are present. Cases with ulceration may display vasculitic changes and prominent upper dermal oedema [110].
LyP type B (lymphocytic or mycosis fungoides-like type) is least common (less than 10%), and features an epidermotropic infiltrate of small to medium-sized lymphoid cells with cerebriform nuclei reminiscent of those seen in MF.
LyP type C (ALCL-like type) consists of a nodular infiltrate of large atypical lymphoid cells arranged in large cohesive sheets with only small numbers of admixed reactive inflammatory cells present.
It should be noted that various histological manifestations of LyP lesions can be seen at the same time in the same patient. The most common association is between LyP types A and C [108].
Immunophenotype.
The tumour cells of LyP types A and C share the same immunophenotype as tumour cells of C-ALCL in their reactivity for CD30 [111,112]. The tumour cells represent a proliferation of activated mature T-helper cells, and usually display reactivity for CD3 and CD4. Activation markers such as HLA-DR [113] and CD25 (interleukin-2 receptor) [99] are also positive. CD7 [113], CD8 [99] and CD56 [114] are negative. Most cases appear to lack CD15 expression, but express cytotoxic molecules such as TIA-1 and granzyme B [99].
In contrast to the expression of CD30 in LyP type A and C cases, the small to medium-sized MF-like tumour cells of LyP type B are usually negative for CD30.
Prognosis and Predictive Factors.
The duration of the disease is variable; most patients have chronic, recurrent crops of lesions for months to years. The significance of this condition is the potential for development of non-lymphoid malignancies (relative risk 3.11) and malignant lymphomas (relative risk 13.33) [115]. Nijsten’s study of 35 childhood cases of LyP also showed a significantly increased risk of developing non-Hodgkin lymphoma as compared to the general population [105]. These data support the practice of life-long monitoring of these patients, even in the absence of skin lesions. Data suggest that immunohistochemical expression of fascin may serve as a prognostic indicator; fascin expression in LyP was significantly less frequent than in ALCL and also than in LyP associated with lymphomas [116].
Treatment.
The treatment options for LyP depend on the degree of skin involvement. Patients with few localized lesions could be clinically observed or treated with topical corticosteroids or immunomodulators. Diffuse lesions may require a more aggressive approach such as PUVA light therapy [117] or systemic medication with low-dose methotrexate [118]. Although all these modalities are helpful in achieving remission, recurrences are noted shortly after therapy discontinuation.
Subcutaneous Panniculitis-Like T-Cell Lymphoma
Definition.
Subcutaneous panniculitis-like T-cell lymphoma (SPTL) is a T-cell lymphoma which preferentially involves the subcutaneous tissue. The category of SPTL in the 2005 WHO-EORTC classification refers specifically to cases with an α/β T-cell phenotype; cases with a γ/δ T-cell phenotype are excluded from this category and included instead in the category of cutaneous γ/δ T-cell lymphomas [1] (see below).
Epidemiology.
Due to the evolving classification of this condition, it is difficult to estimate the exact incidence of this condition.
Clinical Features.
Patients present with acute or recurrent episodes of red-violaceous, deep and tender skin nodules, primarily on the lower extremities or the trunk [119]. The first episode is clinically indistinguishable from other panniculitides. Occasionally, individual nodules may ulcerate. Mild constitutional symptoms may accompany the skin lesions. The episodes last for a few weeks and resolve either spontaneously or with therapy.
Histopathology.
This condition is characterized by a lobular subcutaneous infiltrate composed of an admixture of pleomorphic small, medium to large lymphoid cells. The individual lymphoid cells have rounded nuclei with variably irregular nuclear contours, inconspicuous nucleoli and a moderate amount of pale-staining eosinophilic cytoplasm. The neoplastic lymphoid cells characteristically rim individual adipocytes. Karyorrhexis and necrosis, including fat necrosis, are common. Admixed reactive histiocytes with vacuolated cytoplasm are frequently present, may form a granulomatous reaction and often contain phagocytosed red blood cells and other cellular debris [120]. These are often referred to as ‘bean bag histiocytes’ [121]. Vasculopathic changes range from a pauci-inflammatory thrombogenic vasculopathy to a lymphomatoid vasculitis featuring prominent lymphocytic angio-invasion with mural and luminal fibrin [122]. The lymphoid infiltrate is usually confined to the subcutaneous tissue; focal extension into the overlying lower dermis may be present, but should not be extensive.
Immunophenotype.
The neoplastic lymphoid cells are derived from mature post-thymic cytotoxic T-cells of the adaptive immune system and hence are usually CD3+ and CD8+ with expression of cytotoxic molecules including granzyme B, perforin and T-cell intracellular antigen (TIA-1) [120,123]. CD4 is negative, and there may be aberrant loss of expression of CD5, CD7 and CD3 [40,124,125]. CD30, CD56 and CD57 are negative.
Genetic Features.
The neoplastic T-cells show clonal T-cell receptor gene rearrangements. They are, in the vast majority of cases, negative for Epstein–Barr virus (EBV) sequences [120,123] although EBV genetic material has been found in some cases [126].
Prognosis and Predictive Factors.
Dissemination to lymph nodes and other organs is uncommon, and occurs late in the clinical course of the disease. Patients generally have an indolent waxing and waning clinical course and may achieve remission with combination chemotherapy [127]. A haemophagocytic syndrome may be a complication and usually precipitates a fulminant downhill clinical course.
Treatment.
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


