Usually, accessory tragi are isolated congenital defects. Familial occurrence has been reported. In rare instances, especially if multiple, they are associated with other first branchial arch abnormalities, including cleft lip, cleft palate, branchial fistulae and hypoplasia of the mandible, and are an inconstant feature of the Treacher-Collins, Wolf–Hirschhorn (4p deletion), Townes–Brocks, Delleman and VACTERL (vertebral anomalies, anal atresia, congenital cardiac anomalies, tracheo-oesophageal fistula and/or oesophageal atresia, renal anomalies, radial dysplasia and other limb defects) syndromes. Multiple accessory tragi are a relatively constant finding of the oculo-auriculo-vertebral (Goldenhar) syndrome featuring ear malformations, hemifacial microsomia, epibulbar (lipo)dermoids and vertebral anomalies [1].
The accessory tragus should be differentiated from a macrotragus which shares its histopathological characteristics but represents an enlarged, mostly anteriorly displaced tragus in normal anatomical localization [2].
Treatment.
Accessory tragi can easily be treated by surgical excision. Pedunculated papules may even be removed by ligation or application of a titanium clip flush to the skin. Care must be taken to remove any protuberant portion of the underlying cartilage. Some authors advocate treatment under local anaesthesia in the first days of life.
References
1 Snyder MC, Johnson PJ, Hollins RR. Congenital primary cutis verticis gyrata. Plast Reconstr Surg 2002;110:818–21.
2 Larsen F, Birchall N. Cutis verticis gyrata: three cases with different aetiologies that demonstrate the classification system. Australas J Dermatol 2007;48:91–4.
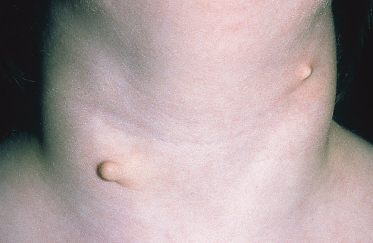
Congenital Cartilaginous Rests of the Neck (Wattles)
Congenital cartilaginous rests of the neck are rare branchiogenic surplus malformations that may be considered the cervical variant of accessory tragi [1]. The synonym ‘wattles’ is derived from the fleshy appendages hanging from the throat of certain birds. The anomaly is always apparent at birth and may be unilateral or bilateral. An irregular, often pedunculated, papule or nodule of ‘springy’, cartilaginous texture is located over or near the lower half of the sternocleidomastoid muscle (Fig. 10.2). Other malformations, such as microtia, stenosis of the external ear canal and branchiogenic fistulae, are rarely associated. Differential diagnosis includes sentinel tags associated with branchial sinuses or fistulae, simple skin tags and benign papillomas. As congenital cartilaginous rests of the neck are not cystic or fistulous; they can easily be excised, which is indicated for cosmetic reasons.
Reference
1 Coras B, Hafner C, Roesch A et al. Congenital cartilaginous rests of the neck (wattles). Dermatol Surg 2005;31:1349–50.
Preauricular Cysts and Sinuses
Aetiology.
Preauricular cysts and sinuses are believed to be the result of an imperfect fusion of the six auditory hillocks of the first two branchial arches. Global prevalence seems to differ considerably (India, 0.17%; USA, 0.1–0.9%; England, 0.9%; China, 1.4%; Taiwan, 1.6–2.5%; some areas of Africa, 4–10%) [1].
Pathology.
The cysts and sinuses are lined by stratified squamous epithelium. The adjacent connective tissue frequently contains skin appendages. After severe inflammation, the lining is often replaced by granulation tissue.
Clinical Features.
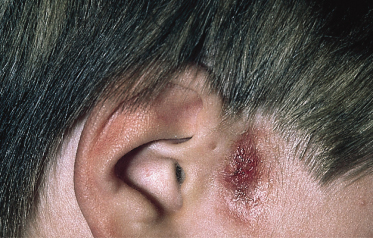
Preauricular sinuses usually present as tiny pits or dells at the anterior margin of the ascending limb of the ear helix (Fig. 10.3). Much less common sites are the lateral surface of the helicine crus and the superior posterior margin of the helix, tragus or lobule [2]. The sinuses may be unilateral, more frequently on the right side, or bilateral, they can be sporadic or inherited. Bilateral occurrence is more likely to be transmitted as an autosomal dominant trait with reduced penetrance and variable expression. The narrow sinuses have a cutaneous mouth and a blind inner end, almost always connecting to the perichondrium of the auricular cartilage. They may arborize, with a subcutaneous network of cysts arranged around their tracts, and rarely extend into the parotid gland.
In 3–10% of cases, they are associated with deafness and with several complex developmental defects, branchio-oto-renal (BOR) syndrome and branchio-otic (BO) syndrome being the by far most important. BOR and BO syndromes are autosomal dominant conditions characterized by preauricular pits, conductive, sensorineural or mixed hearing loss, structural defects of the outer, middle or inner ear, lateral cervical fistulas, cysts or sinuses, and nasolacrimal duct stenosis or fistulas. Renal anomalies and failure are an additional feature of BOR syndrome [1,2]. Both syndromes are frequently caused by mutations in the human homologue of the Drosophila eye absent (EYA1) gene. Therefore, auditory testing and renal ultrasound is regarded as imperative if a preauricular sinus is accompanied by one or more of the following: other malformations or dysmorphic features, a family history of deafness, auricular and/or renal malformations or a maternal history of gestational diabetes [3].
In most cases preauricular sinuses remain asymptomatic, at least until adulthood [1]. However, the seemingly trivial and often unnoticed anomaly can give rise to troublesome symptoms if infection supervenes (Fig. 10.3). In this case, recurrent purulent discharge and preauricular abscesses causing oedema, erythema and pain may develop.
Sonography is helpful to confirm the diagnosis and to determine the extent of the sinus and cysts and their relation to adjacent anatomical structures.
Treatment.
If asymptomatic a preauricular sinus does not require intervention. Cure of symptomatic sinuses is attained only by meticulous complete excision of the entire tract and its associated cysts. This may be difficult to ensure, especially in the presence of infection. Thus, most authors recommend surgery only after pathogen-targeted antibiotic treatment of the infection. Nevertheless, postoperative recurrences are reported to occur in 9–42% of cases. General anaesthesia, magnification employed during surgery, wide local excision with removal of cartilage at the base of the tract, demonstrating the sinus with methylene blue injection and probing at the time of surgery help to reduce the recurrence risk.
References
1 Huang XY, Tay GS, Wansaicheong GK, Low WK. Preauricular sinus: clinical course and associations. Arch Otolaryngol Head Neck Surg 2007;133:65–8.
2 Scheinfeld NS, Silverberg NB, Weinberg JM, Nozad V. The preauricular sinus: a review of its clinical presentation, treatment, and associations. Pediatr Dermatol 2004;21:191–6.
3 Wang RY, Earl DL, Ruder RO, Graham JM Jr. Syndromic ear anomalies and renal ultrasounds. Pediatrics 2001;108:E32.
Branchial Cysts, Sinuses and Fistulae
Aetiology.
Branchial cysts, sinuses and fistulae result from maldevelopment of the branchial apparatus, most often the second branchial arch, during the embryological period. Familial occurrence has repeatedly been reported.
Pathology.
The sinus tracts of branchial anomalies are lined by stratified squamous epithelium, which may in part be replaced by respiratory (ciliated columnar) epithelium. There is often abundant subepithelial lymphoid tissue. Thymic or thyroid tissue points to derivation from the third or fourth branchial arch, respectively. The presence of smooth muscle, cartilage and seromucous glands in branchial arch remnants may lead to confusion with a bronchogenic cyst (see below).
Clinical Features.
External branchial sinuses and fistulae are detectable as openings located on the lateral neck, at the anterior border of the sternocleidomastoid muscle. Sinuses end blindly in the deep neck tissue, while fistulae have a continuous communication between skin and mucosa and may drain into the tonsillar area of the pharynx. Cysts present as painless mobile swellings and have neither a cutaneous nor a mucosal opening. The external orifice of sinuses and fistulae may exude a mucous secretion. Early secondary infection is frequent and may cause significant morbidity. Because of their obvious skin opening and associated drainage and infection, branchial sinuses and fistulae are usually diagnosed much earlier than branchial cysts, most often in the first few years of life [1].
Bilateral lesions are rare; preauricular pits may be an associated finding. Branchial anomalies may form part of complex hereditary conditions, such as BOR syndrome, branchio-oculo-facial syndrome and others.
Diagnosis of branchial sinuses and fistulae is obvious in light of the cutaneous opening in typical location. In contrast, branchial cysts are part of the extensive differential diagnosis of lateral neck masses including lymphadenitis, abscess, lipoma, cystic hygroma, haemangioma, lymphangioma, lymphoma, ectopic thyroid cyst, thyroglossal duct cyst and cervical thymic cyst [1,2]. Radiographical imaging, particularly CT scans, ultrasound and colour flow Doppler help in discrimination and assessment of extension.
Treatment.
As there is no spontaneous regression of branchial anomalies and complication by recurrent infections is frequent, complete surgical removal is the treatment of choice. Ideally, surgery is performed under general anaesthesia at 1 year of age before infections occur.
References
1 Schroeder JW Jr, Mohyuddin N, Maddalozzo J. Branchial anomalies in the pediatric population. Otolaryngol Head Neck Surg 2007;137:289–95.
2 Acierno SP, Waldhausen JH. Congenital cervical cysts, sinuses and fistulae. Otolaryngol Clin North Am 2007;40:161–76, vii–viii.
Congenital Midline Cervical Cleft
This rare congenital anomaly, most likely related to aberrant fusion of the first or second branchial arches, is located in the anterior midline of the neck at any point between the mandible and the sternum. The characteristic clinical presentation consists of a cephalic nipple-like projection (‘skin tag’) with the inferior margin being formed by a short sinus of about 1 cm length and an atrophic mucosal surface dividing the two [1]. An underlying fibrous cord may limit the movement of the neck and cause webbing. The cleft is covered by stratified squamous epithelium lacking skin appendages while the sinus tract is usually lined by pseudo-stratified columnar epithelium and often contains seromucinous salivary glands [2]. The abnormality may be isolated or associated with a broad spectrum of midline defects, such as median cleft of the mandible, tongue and lower lip. Complete excision of the anomaly and the underlying fibrous tract within the first year of life is critical to avoid scarring contracture and mandibular deformities. The surgical defect is usually closed with multiple Z-plasties.
References
1 Gardner RO, Moss AL. The congenital cervical midline cleft: case report and review of literature. Br J Plast Surg 2005;58:399–403.
2 Agag R, Sacks J, Silver L. Congenital midline cervical cleft. Cleft Palate Craniofac J 2007;44:98–101.
Thyroglossal Duct Cysts
Thyroglossal duct cysts develop as a result of mucus production into an incompletely obliterated thyroglossal duct. Most often, they lie close to the hyoid bone in or near the midline of the neck, but they may be located at any site along the pathway of the thyroid anlage. They are the most common cause of midline neck swellings in childhood and adolescence although a significant minority of cysts is diagnosed not before adulthood. Typically, a painless soft mass of 1–2 cm diameter moves upwards on swallowing and on protrusion of the tongue. Less often, there may be a discharging sinus occurring spontaneously or after insufficient operation. Infection is a problem increasingly frequent with age. To avoid this and malignant change, thyroglossal duct cysts should be treated with an early Sistrunk’s operation, which includes resection of the mid-portion of the hyoid bone and en bloc cystectomy with a cuff of muscle around the duct to the base of the tongue. Postoperative recurrence rates are in the range of 3–4%. Prior to surgery, the normal thyroid gland should be identified by an imaging modality. On ultrasound, the cysts themselves show an unechoic, hypoechoic, pseudo-solid or heterogeneous pattern [1]. Neck computed tomography, MRI, fine-needle aspiration and radioisotope thyroid scanning offer more exact information about location, size and relation to adjacent structures and help to exclude differential diagnoses, such as ectopic thyroid tissue, lipoma, lymphadenopathy and carcinoma but are not required as a matter of routine [2]. Histologically, the ducts are frequently multiple and branched. Numerous cases of malignancy arising in thyroglossal duct remnants have been reported, the thyroid papillary carcinoma being by far the most common type of neoplasia.
References
1 Kutuya N, Kurosaki Y. Sonographic assessment of thyroglossal duct cysts in children. J Ultrasound Med 2008;27:1211–19.
2 Lin ST, Tseng FY, Hsu CJ, Yeh TH, Chen YS. Thyroglossal duct cyst: a comparison between children and adults. Am J Otolaryngol 2008;29:83–7.
Cutaneous Bronchogenic Cysts
The precise aetiology of cutaneous bronchogenic cysts is disputed. The more common cysts inside the thoracic cage are believed to originate from accessory buds that became sequestrated from the primitive tracheobronchial tree or primitive foregut. This concept also fits for presternal cysts that may develop after intervention of fusing sternal bars, but it is unlikely for bronchogenic cysts in ectopic locations, such as the shoulder and scapular regions. Transplantation via lymphatic or haematogenous routes, metaplasia and maldifferentiation of epithelial components to ciliated epithelium are other possibilities.
In most cases, the lesion is noted at or shortly after birth as a swelling or draining sinus on the lower neck, the anterior upper chest near the suprasternal notch or over the manubrium sterni. Chin, shoulder, scapular region, back and abdomen are less common sites [1]. There is no connection to underlying structures. Males are predominantly affected. Histological examination of bronchogenic cysts typically reveals lining of ciliated and mucin-producing pseudostratified columnar epithelium of respiratory type.
Excision is recommended to prevent infection and malignant transformation.
Reference
1 Ozel SK, Kazez A, Koseogullari AA, Akpolat N. Scapular bronchogenic cysts in children: case report and review of the literature. Pediatr Surg Int 2005;21:843–5.
Skin Dimples
Skin dimples are unilateral or more often bilateral deep cutaneous depressions commonly seen over bony prominences, such as the acromial process of the scapula, os sacrum, elbow, patella and tibia. They are thought to be caused by early entrapment-induced fixation of the skin to the underlying bony prominence, thus preventing the development of subcutaneous tissue.
Usually, skin dimples are a congenital isolated finding and, as in the case of bilateral acromial or shoulder dimpling, also known as ‘supraspinous fossae’, may represent a harmless autosomal dominant anomaly [1]. However, bilateral acromial dimples have also been reported as a relatively constant feature of the 18q deletion syndrome, and as an occasional finding in trisomy 9p syndrome, Russell–Silver dwarfism and Say syndrome [2]. Recently, an association of subacromial dimples with recurrent posterior dislocation of the shoulder has been postulated. Dimples overlying the tibia are seen in camptomelic dwarfism, oral–facial–digital syndrome and osteoglophonic dysplasia. Dimples over elbows and knees have been observed in prune belly syndrome, Joubert syndrome and facial clefting syndrome. Midline sacral dimples are an inconstant minor feature of various congenital malformation syndromes. If large, more than 2.5 cm away from the anus or combined with other lesions, they may be associated with occult spinal dysraphism. Sporadic cases of skin dimpling have been described after congenital rubella, hypophosphatasia and misoprostol exposure in utero. Dimple-like scars are not uncommon after amniocentesis.
References
1 Beillard C, Guillet G, Vabres P, Dagregorio G, Larregue M. Bi-acromial dimples: a series of seven cases. Pediatr Dermatol 2005;22:412–14.
2 Virgili A, Tosti G, Bettoli V, Corazza M. Multiple congenital symmetric skin dimples. Dermatology 2002;204:293–5.
Transverse Nasal Line
The transverse nasal line runs transverly across the skin between the upper two-thirds and lower third of the nose. The clinical appearance varies from a faint reddish line to a more visible groove or crease with a depth and width of up to several millimetres [1]. Particularly in Afro-Caribbeans, it may be hyperpigmented. It appears early in life and often remains undefinitely. There is a clear familial predisposition, and females seem to be affected more often.
Based on its special location, the transverse nasal line is presumed to represent an anatomical variant resulting from differential enlargement of the alar and triangular cartilages of the nose. Of note, it is a localizing factor for milia, cysts and comedones. Inflammation may lead to ‘pseudo-acne of the nasal crease’ [2].
The transverse nasal line has to be distinguished from the nasal crease or wrinkle developing in patients with allergic rhinitis as a response to frequent rubbing the nose upward for relief of itching (the ‘allergic salute’).
References
1 Shelley WB, Shelley ED, Pansky B. The transverse nasal line: an embryonic fault line. Br J Dermatol 1997;137:963–5.
2 Risma KA, Lucky AW. Pseudoacne of the nasal crease: a new entity? Pediatr Dermatol 2004;21:427–31.
Congenital Lip Pits
Congenital lip pits can be divided into three types: commissural pits, midline sinuses of the upper lip and lower lip pits (lip sinuses).
Commissural pits are the most common type of lip pits, found in about 0.5–2% of neonates. They are located within the oral cavity at the angle of the mouth and represent the openings of bilateral, blind-ending sinuses of short length. They are frequently inherited as an isolated autosomal dominant anomaly, but may be, as a feature of the BOR syndrome, associated with branchial, hearing and renal anomalies. Furthermore, commissural lip pits have been described in association with alveolar synechia, ankyloblepharon filiforme adnatum and ectodermal defects.
In the midline sinus of the upper lip, the opening of a blind-ending sinus tract is seen in the midline of the upper-lip philtrum. Hypertelorism, nasal fistula and lip fistula may be associated.
Congenital lower-lip pits result from a fusion defect of the lower part of the first branchial arch. They usually present as bilateral symmetrical depressions on the vermilion portion of the lower lip on each side of the midline. Occasionally, the ostia are surrounded by conical protrusions. The orifices represent the end points of blind sinuses that extend inward into the orbicularis oris muscle to a depth of up to 20 mm or more and may bifurcate. The tracts are lined by stratified squamous epithelium. When they communicate with the ducts of the underlying minor salivary glands, saliva or mucus may be discharged from the otherwise asymptomatic openings. Occasionally, only a single paramedian or median sinus may be present. Labial conical elevations without fistulous ostia are believed to represent the microform of the anomaly [1].
Congenital lower-lip pits are the hallmark of van der Woude syndrome, an autosomal dominant craniofacial disorder with high penetrance and variable expressivity characterized by lower-lip pits and cleft lip, cleft palate or both. Its prevalence is indicated with 1 in 40,000 to 1 in 100,000 births. Other inconstant features include hypodontia, gingival synechiae, ankyloglossia, syndactyly of the hands, thumb hypoplasia, foot deformities, symblepharon, preauricular sinuses, polythelia, congenital heart disease and cerebral abnormalities [2]. In about two-thirds of patients, congenital lower-lip pits are the only manifestation of the syndrome. They are also a characteristic feature of the popliteal pterygium syndrome (popliteal webs, cleft lip and/or cleft palate, and anomalies of the genitourinary system). Van der Woude syndrome and popliteal pterygium syndrome are allelic conditions caused by diverse mutations of the gene encoding the interferon regulatory factor 6 (IRF6) [3]. Therefore, genetic counselling has to respect the considerable phenotypic variation and overlap of the two conditions. Furthermore, lower-lip pits are a facultative sign of the orofaciodigital syndrome type 1 and the Kabuki make-up syndrome.
The only treatment for symptomatic or unpleasant lip pits is surgical excision ensuring complete removal of the often bifurcating tracts.
References
1 Rizos M, Spyropoulos MN. Van der Woude syndrome: a review. Cardinal signs, epidemiology, associated features, differential diagnosis, expressivity, genetic counselling and treatment. Eur J Orthod 2004;26:17–24.
2 Dissemond J, Haberer D, Franckson T, Hillen U. The van der Woude syndrome: a case report and review of the literature. J Eur Acad Dermatol Venereol 2004;18:611–13.
3 Ziai MN, Benson AG, Djalilian HR. Congenital lip pits and van der Woude syndrome. J Craniofac Surg 2005;16:930–2.
Supernumerary Nipples (Polythelia)
With a prevalence of 0.2–5.6% in different series, supernumerary nipples or polythelia are the most frequently encountered congenital abnormality of the breast. They are remnants of the embryological milk line which runs from the anterior axillary fold to the inner thigh. Most often, they are located on the chest wall and upper abdomen and present as small brown or pink, umbilicated or elevated papules surrounded by a pigmented areola (Fig. 10.4) [1]. A nipple may also be seen without areola, and vice versa. Usually, there is only a single lesion, but multiple and bilateral nipples are possible. In a prospective study, prevalence was higher on the left side and in males.
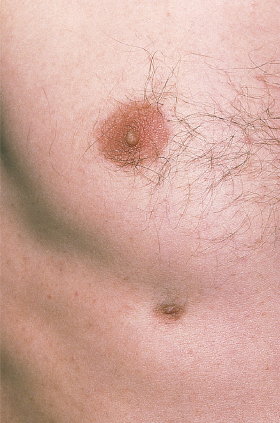
Familial occurrence without further anomalies suggesting autosomal dominant transmission has been observed. Polythelia forms part of the X-linked Simpson–Golabi–Behmel syndrome and another syndrome additionally including aplasia cutis congenita, microcephaly and developmental delay with apparent autosomal dominant inheritance. It has also been repeatedly observed in association with Becker naevus.
Sporadic accessory nipples seem to be associated with malformations of the urinary tract, in particular the kidneys, varying in frequency from 1.2% to 27% [2]. This relationship has not stood uncontradicted. Furthermore, an association of supernumerary nipples with haematological disorders and congenital heart anomalies has been postulated. Remarkably, mammary Paget disease can arise in a supernumerary nipple.
Accessory nipples are occasionally mistaken for melanocytic naevi [3]. They should also be differentiated from unilateral or bilateral intra-areolar polythelia with two or more nipples located within the areola, caused by intrauterine division (dichotomy) of the developing nipple. Excision may be indicated for diagnostic or cosmetic reasons.
References
1 Brown J, Schwartz RA. Supernumerary nipples: an overview. Cutis 2003;71:344–6.
2 Ferrara P, Giorgio V, Vitelli O et al. Polythelia: still a marker of urinary tract anomalies in children? Scand J Urol Nephrol 2009;43:47–50.
3 Latham K, Fernandez S, Iteld L et al. Pediatric breast deformity. J Craniofac Surg 2006;17:454–67.
Other Abnormalities of the Breast and Nipple
Accessory breast tissue (polymastia) may underlie a supernumerary nipple and is usually noted only after hormonal stimulation at puberty or in pregnancy. Supernumerary breasts are located along the primitive milk line with the axilla being a particularly common site where they can be mistaken for lymphadenopathy or lipoma. The prevalence seems to be higher in women and those of Oriental extraction. As polythelia, polymastia can be associated with other, especially renal, anomalies. Fine-needle aspiration may serve as a useful tool for diagnosis. Complete excision of ectopic breast tissue is recommended because of the risk of malignant transformation [1].
Athelia is defined as complete absence of the nipple–areola complex and may be unilateral or bilateral. It is generally noted at birth or shortly thereafter. The rare anomaly is almost always combined with amastia (absence of the breast and nipple) within the frame of syndromic disorders of development. These include ectodermal dysplasia, Al Awadi–Raas-Rothschild syndrome (absence or severe hypoplasia of skeletal parts of the limbs), Poland syndrome (see below), choanal atresia–athelia syndrome, scalp–ear–nipple syndrome and ADULT (acro-dermato-ungual-lacrimal-tooth) syndrome [2]. An isolated case of unilateral athelia was associated with an underlying dermoid cyst. Athelia is believed to be caused by a failure in parathyroid hormone-related protein production.
Unilateral absence (amastia) or hypoplasia (hypomastia) of the breast and nipple are most often a feature of the Poland syndrome. This congenital anomaly consists of a spectrum of congenital deformities of the chest wall, breast and upper extremity. Unilateral absence of the sternocostal head of the pectoralis major muscle is the indispensable prerequisite for diagnosis. In addition, amastia to mild hypoplasia of the breast, athelia or abnormal nipple, scarcity of subcutaneous tissue over the pectoral region, absence of the pectoralis minor muscle, deficiency of additional chest wall muscles, aplasia and/or deformity of costal cartilages or anterior ribs II–V, alopecia of axillary and mammary regions, and ipsilateral hand anomalies (brachysyndactyly, syndactyly, hypoplasia of middle phalanges) may be present [1]. Less frequent associated defects include renal malformations, dextrocardia and vertebral abnormalities. Poland syndrome is thought to be caused by interruption of the embryonic blood supply to the upper limb bud in the sixth week of gestation with consecutive hypoplasia of the subclavian artery. It occurs in about 1 out of 25,000 live births. Men are affected three times more often than females, and the right side three times more often than the left [3]. Most cases are sporadic, but paradominant inheritance has been proposed to explain the rare familial cases. Evaluation of the vascular status is mandatory prior to reconstructive breast surgery.
Hypomastia is a feature of the Becker naevus syndrome and has been observed in conjunction with naevus depigmentosus and phacomatosis cesioflammea in individual cases.
While functional deficits in breast and nipple deformities are corrected early, aesthetic surgery is postponed until after puberty to achieve the maximum possible symmetry [1].
References
1 Latham K, Fernandez S, Iteld L et al. Pediatric breast deformity. J Craniofac Surg 2006;17:454–67.
2 Ishida LH, Alves HR, Munhoz AM et al. Athelia: case report and review of the literature. Br J Plast Surg 2005;58:833–7.
3 Borschel GH, Costantino DA, Cederna PS. Individualized implant-based reconstruction of Poland syndrome breast and soft tissue deformities. Ann Plast Surg 2007;59:507–14.
Developmental Abnormalities of the Umbilicus
A variety of congenital umbilical lesions result from complete or partial failure of obliteration of two embryological structures: the omphalomesenteric (vitelline) duct and the urachus. The clinical features depend upon the site and extent of patency of these structures.
A completely patent omphalomesenteric duct is exceedingly rare and leads to a fistula between ileum and umbilicus. The anomaly is noted soon after birth by faecal discharge severely irritating the adjacent skin. Sudden ileal prolapse through the umbilicus may complicate the anomaly and represents a surgical emergency because of the danger of strangulation and necrosis of the affected bowel.
A partially patent omphalomesenteric duct gives rise to a umbilical polyp of bright red colour (remnant of the peripheral portion) [1,2], an umbilical sinus (patency of the peripheral portion) or a vitelline cyst (patency of the intermediate portion). The latter is usually asymptomatic whereas polyps and sinuses mostly secrete an exudate of serous or mucoid, more rarely of bloody or serosanguinous, character from birth or shortly afterwards. This distinguishes the umbilical polyp from the most important differential diagnosis, granuloma pyogenicum, which usually has no or a purulent secretion. Polyps and cysts are frequently accompanied by potentially serious internal omphalomesenteric remnants, such as Meckel’s diverticulum attached to the umbilicus by obstructing fibrous bands.
A completely patent urachus leads to fistula between the urinary bladder and umbilicus. It presents soon after birth by leakage of urine from the abnormal-appearing umbilicus, which may cause irritation of periumbilical skin. An umbilical tumour which may be confused with pyogenic granuloma and infection of the genitourinary tract may occur.
A partially patent urachus may result in an umbilical urachal sinus (patency of the peripheral portion) or an urachal cyst (patency of the intermediate portion). Unless large or infected, they present as asymptomatic cystic swellings. Adenocarcinoma of the urachus may rarely occur in later life and has a poor prognosis.
The diagnosis of developmental anomalies can be verified by radiographical imaging, especially ultrasound and sinography, and histological demonstration of ectopic gastrointestinal or bladder transitional epithelium covering polyps or lining sinuses and cysts.
Umbilical enteric and urinary fistulae are absolute indications for surgical intervention [3]. Simple excision of umbilical polyps should be performed only if underlying intestinal and urinary tract anomalies are ruled out [1]. More often, the lesion has to be completely excised together with the attached part of the bladder or bowel. Total resection of the umbilicus is usually not required.
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


