14 Wound healing
Synopsis












Introduction
Physical trauma represents one of the most primitive challenges that threatened survival. In other words, injury eliminated the unfit. A Sumerian clay tablet (c. 2150 bc) described early wound care that included washing the wound in beer and hot water, using poultices from substances such as wine dregs and lizard dung, and bandaging the wound. Ancient scriptures depicting the science of life or Ayurveda date from the sixth to seventh century bc, and represent the beginning of planned physical injury with the intent to cure.1,2 Hippocrates (c. 400 bc) detailed the importance of draining pus from the wound, and Galen (c. 130–200 ad) described the principle of first- and second-intention healing.3 Wound healing advanced slowly over the centuries, with major advances in the 19th century in the importance of controlling infection, hemostasis, and necrotic tissue.4 Today, surgical trauma, taken together with injury caused during accidents and secondary to other clinical conditions, e.g., diabetes, represents a substantial cost to society.5 Any solution to wound-healing problems will require a multifaceted comprehensive approach. First and foremost, the wound environment will have to be made receptive to therapies. Second, the appropriate therapeutic regimen needs to be identified and provided while managing systemic limitations that could secondarily limit the healing response. This chapter aims to present an overall outline of the cutaneous wound-healing process.
Acute wounds
Open wounds may be generally categorized as:
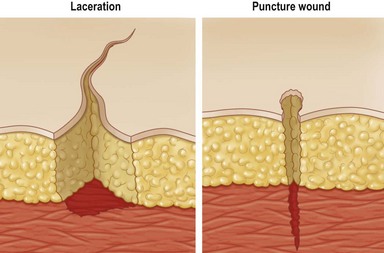
• Lacerations – ragged tears and cuts; masses of torn tissue underneath; caused by dull knife, bomb fragments and machinery and may include crushing of tissues; frequently contaminated (Fig. 14.1)
• Puncture – e.g., sharp penetrations caused by nails, needles, wire, or bullets (Fig. 14.1). These are of great concern in patients with diabetes, as many of these patients have polyneuropathy and have insensate feet, leading to occult injury. Many times these patients will step on thumb tacks, safety pins, or other sharp household objects and not even know it: this, coupled with compromised vascular status, leads to chronic wound infection
• Abrasions – the superficial layer of the skin is removed; skinned knee or elbows and rope burns are examples; an abrasion lends itself to infection
• Avulsions – sections of skin torn off either in part (attached to body) or totally (detached from body); heavy bleeding is common
• Amputations – traumatic amputation results in nonsurgical removal of limb from the body and accompanies heavy bleeding.
There are three general techniques of wound treatment:
1. primary intention, in which all tissues, including the skin, are closed with suture material after completion of the operation
2. secondary intention, in which the wound is left open and closes naturally
3. tertiary intention, in which the wound is left open for a number of days and then closed if it is found to be clean.
The wound-healing process
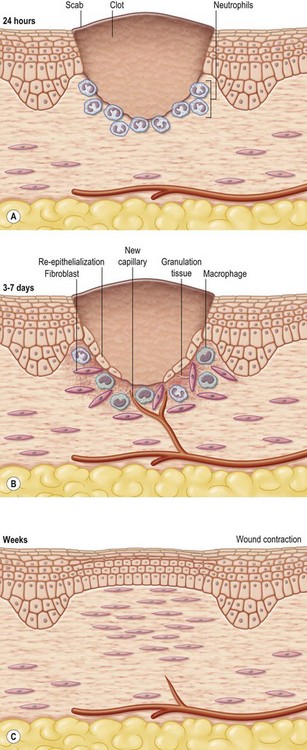
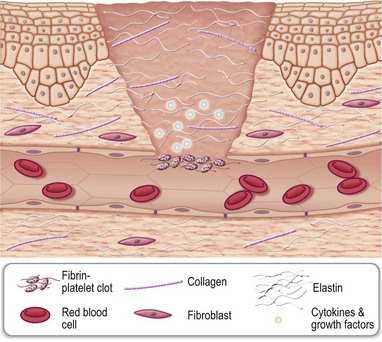
The entire wound-healing process may be viewed as a cascade that is governed by numerous feedback and feedforward regulatory loops driven by signals from the wound tissue itself, wound microenvironment, as well as interventions under conditions where the wound is subjected to therapy. For simplicity of understanding the several interdigitated biological processes that drive the overall healing response, wound healing is commonly discussed as the following overlapping phases: hemostasis and inflammation, proliferative (granulation, vascularization, and wound closure; closure may be discussed as wound contraction and epithelialization) and remodeling (can continue from weeks to years and encompasses scarring, tensile strength, and turnover of extracellular matrix (ECM) components). These stages, taken as a whole, are also referred to as the wound-healing cascade (Fig. 14.2).
Hemostasis
For bleeding wounds, the highest priority is to stop bleeding and this is achieved by hemostasis. Hemostasis is thus a protective physiological response to vascular injury that results in exposure of blood components to the subendothelial layers of the vessel wall. Through successful hemostasis blood loss is prevented by plugging the wound within seconds through vasoconstriction and formation of a hemostatic blood clot consisting of platelets and fibrin. Hemostasis requires both platelets and the blood coagulation system. The process of blood coagulation may be subdivided into initiation and amplification. Initiation is caused by an extrinsic pathway, whereas amplification is executed by an intrinsic pathway. The intrinsic pathway consists of plasma factor XI (FXI), IX, and VIII (Fig. 14.3). Tissue factor (TF) generates a “thrombin burst,” a process by which thrombin is released instantaneously. Thrombin is a key driver of the overall coagulation cascade.
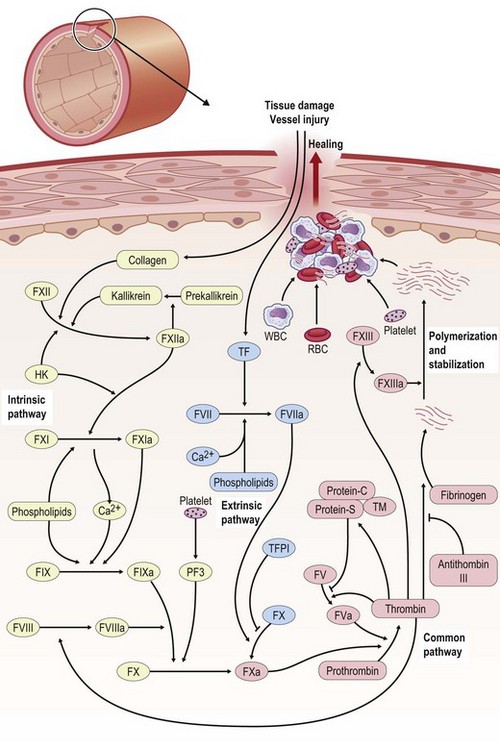
The extrinsic pathway responsible for the initiation of blood coagulation consists of the transmembrane receptor TF and plasma FVII/VIIa. On the other hand, the intrinsic pathway consists of plasma FXI, FIX, and FVIII. Under physiological conditions, TF is constitutively expressed by adventitial cells surrounding blood vessels and initiates clotting. Examples of such adventitial cells include vascular smooth-muscle cells, pericytes, and adventitial fibroblasts.6,7 TF may also contribute to amplification of blood coagulation through its so-called blood-borne form, which is present as cell-derived microparticles as well as through TF expressed within platelets.6–8
Levels of FVIIa, a key player of the extrinsic pathway, in the circulating blood are higher than any other activated coagulation factor. Following injury to the blood vessel wall, FVII comes into contact with TF expressed on TF-bearing cells (e.g., white blood cells) and forms an activated complex (TF–FVIIa). TF–FVIIa activates FIX and FX, resulting in FIXa and FXa, respectively. FVII is activated by thrombin, FXIa, FXII, and FXa. The activation of FXa by TF–FVIIa is almost immediately inhibited by the TF pathway inhibitor. FXa and its cofactor FVa form the prothrombinase complex, which activates prothrombin to thrombin. Thrombin is a serine protease that plays a central role in hemostasis after tissue injury by converting soluble plasma fibrinogen into an insoluble fibrin clot and by promoting platelet aggregation. Thrombin activates other components of the coagulation cascade, including FV and FVIII, which in turn activates FXI and cascades to the activation of FIX. Thrombin also activates and releases FVIII from being bound to von Willebrand factor. FVIIIa is the cofactor of FIXa, and together they form the “tenase” complex, which activates FX. In this way the cycle continues. The intrinsic pathway begins with formation of the primary complex on collagen by high-molecular-weight kininogen, prekallikrein, and FXII (Hageman factor). Prekallikrein is converted to kallikrein, and FXII becomes FXIIa. FXIIa converts FXI into FXIa. FXIa activates FIX, which together with its cofactor FVIIIa forms the tenase complex. The tenase complex activates FX to FXa (Fig. 14.3).
The blood clot physically helps plug the wound, minimizing blood loss. It is primarily made up of cross-linked fibrin, cells such as erythrocytes and platelets, as well as other ECM proteins such as fibronectin, vitronectin, and thrombospondin. Current understanding portrays the clot as a dynamic structural matrix containing functionally active proteins and cells. In addition to containment of blood loss, the clot serves as a first aid against microbial invasion. The clot also serves as a provisional matrix for the homing of blood-borne cells, including inflammatory as well as stem or progenitor cells. The provisional matrix is enriched in cytokines and growth factors which then regulate the function of the homing cells.9,10
The formation of blood clot is initiated by the proteolytic cleavage of fibrinogen by thrombin. As a result, fibrin is produced and forms cross-links with each other. Cross-linked fibrin entraps platelets, and together they adhere to the subendothelium through adhesion molecules called integrins. Clot fibrin plays a key role in mounting the inflammatory process as well as in facilitating wound angiogenesis and stromal cell proliferation. Fibrin binds to integrin CD11b/CD18 on infiltrating monocytes and neutrophils. It also binds to fibroblast growth factor-2 (FGF-2) and vascular endothelial growth factor (VEGF) that help the wound tissue vascularize. Fibrin also binds to insulin-like growth factor-I and promotes stromal cell proliferation.11,12
Blood clot represents the seat of wound chemotaxis. Thrombin, released by platelets at the wound site, is an early mediator of clot development.13 Thrombin is a serine protease that converts soluble plasma fibrinogen into an insoluble fibrin clot. In addition, it promotes platelet aggregation. Thrombin function may be viewed as an interface between the hemostasis phase of wound healing and the ensuing inflammatory phase as it plays a potent role in mounting wound inflammation. The proinflammatory effects of thrombin include stimulation of vasodilation responsible for plasma extravasation, edema, and an increased expression of endothelial cell adhesion molecules that helps monocytes and others cells extravasate and infiltrate the wound site. Thrombin also induces the release of proinflammatory cytokines like CCL2, interleukin-6 (IL-6), and IL-8 by endothelial cells. These cytokines induce monocyte chemotaxis.14 Furthermore, thrombin induces the release of inflammatory cytokines by monocytes, including IL-6, interferon-γ, IL-1β, and tumor necrosis factor-α (TNF-α). These early-phase cytokines are typically proinflammatory, which may govern the differentiation of blood-derived monocytes into M1 wound macrophages.15 Wound chemotaxis is also driven by the degradation of fibrin and subsequent activation of the complement system. As part of this process several chemotactic agents and cytokines are released, which in turn launch the inflammatory phase of wound healing through chemotactic recruitment of blood-borne immune cells.16 Platelets are one of the earliest sources of cytokines which execute immune cell chemotaxis as well as macrophage activation. Once trapped in the fibrin net, platelets release granules that function as a reservoir for biologically active proteins, such as RANTES (regulated on activation, normal T cell expressed, and secreted or CCL5), thrombin, transforming growth factor-β (TGF-β), platelet-derived growth factor (PDGF), and VEGF. CCL5 is one of the most potent monocyte chemoattractants released by platelets after injury. Other cytokines and chemokines that attract monocytes to the wound bed include monocyte chemoattractant protein-1 (MCP-1) (CCL2), MIP-1α (CCL3), TGF-α, fibronectin, elastin, C5a, C3a, nerve growth factor, and ECM components.17,18
Inflammation
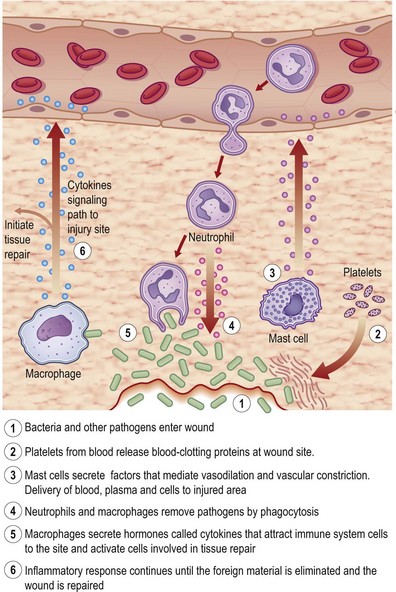
Tissue injury triggers an acute-phase inflammation (Latin, inflammare, to set on fire) response that is meant to prepare the wound site for subsequent wound closure (Fig. 14.4). Inflammation encompasses a series of responses of vascularized tissues of the body to injury. Local chemical mediators that are biosynthesized during acute inflammation give rise to the macroscopic events characterized by Celsus in the first century, namely, rubor (redness), tumor (swelling), calor (heat), and dolor (pain). At cellular and molecular levels, inflammation results from the coordination of manifold systems of receptors and sensors that affect transcriptional and posttranslational programs necessary for host defense and resolution of infection. During normal healing, the inflammatory response is characterized by spatially and temporally changing patterns of specific leukocyte subsets.
Platelets
Formation of the clot or thrombus is dependent on platelet activation. The platelet-rich blood clot also entraps polymorphonuclear leukocytes (neutrophils). This helps amplify blood coagulation and lays the foundation for the subsequent acute-phase inflammatory response. In a matter of hours after injury, large numbers of neutrophils extravasate by transmigrating across the endothelial cell wall of blood capillaries to the wound site. To enable this, local blood vessels are activated by proinflammatory cytokines such as IL-1β, TNF-α, and interferon-γ. These cytokines induce the expression of adhesion molecules necessary for adhesion of leukocytes and diapedesis (Fig. 14.5). Adhesion molecules such as integrins as well as P-selectin and E-selectin play a central role in enabling diapedesis of neutrophils (Fig. 14.6). These adhesion molecules bind with integrins expressed on the cell surface of neutrophils, such as CD11a/CD18 (LFA), CD 11b/CD18 (MAC-1), CD11c/CD18 (gp150, 95), and CD11d/CD18. Alongside cytokines, chemokines play a major role in mounting acute-phase inflammation after injury. Chemokines include IL-8, MCP-1, and growth-related oncogene-α. In the case of an infected wound, bacterial products such as lipopolysaccharide and formyl-methionyl peptides can enhance neutrophil recruitment to the wound site (Fig. 14.7).

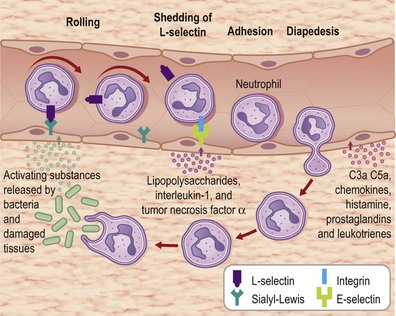
Neutrophils
Neutrophils traverse postcapillary venules at sites of inflammation, degrade pathogens within phagolysosomes, and undergo apoptosis. Neutrophils serve a wide range of functions, ranging from phagocytosis of infectious agents to cleansing of devitalized tissue. When coated with opsonins (generally complement and/or antibody), microorganisms bind to specific receptors on the surface of the phagocyte and invagination of the cell membrane occurs with the incorporation of the microorganism into an intracellular phagosome. There follows a burst of oxygen consumption, and much, if not all, of the extra oxygen consumed is converted to highly reactive oxygen species. This is called respiratory burst. In addition, the cytoplasmic granules discharge their contents into the phagosome, and death of the ingested microorganism soon follows. Among the antimicrobial systems formed in the phagosome is one consisting of myeloperoxidase (MPO), released into the phagosome during the degranulation process, hydrogen peroxide (H2O2), formed by the respiratory burst and a halide, particularly chloride. The initial product of the MPO-H2O2-chloride system is hypochlorous acid, and subsequent formation of chlorine, chloramines, hydroxyl radicals, singlet oxygen, and ozone has been proposed. These same toxic agents can be released to the outside of the cell, where they may attack normal tissue and thus contribute to the pathogenesis of disease.19 Other products delivered by neutrophils to the wound site include antimicrobials such as cationic peptides and eicosanoids as well as proteases such as elastase, cathepsin G, proteinase 3, and urokinase-type plasminogen activator. As it relates to the overall inflammatory process elicited in response to injury, neutrophils are major players because they can modify macrophage function and therefore regulate innate immune response during wound healing.20 In the absence of neutrophils, wound site macrophages seem to lack guidance in conducting the healing process.21
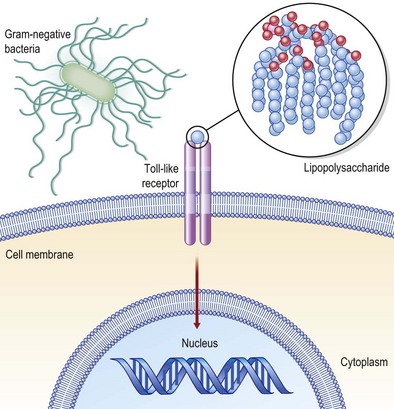
Mediators present in the microenvironment that the monocyte traverses to reach the wound site interact with receptors on the monocyte cell surface, bringing forth major changes in the transcriptomic as well as proteomic make of the cell. Major examples of such receptors present on the monocyte surface include Toll-like receptors (TLRs: Fig. 14.8), complement receptors, and Fc receptors. At the wound site, macrophages function as antigen-presenting cells and phagocytes scavenging dead cells and debris. In addition, they deliver a wide range of growth factors that are known for their abilities to execute the wound-healing process. Such growth factors include TGF-β, TGF-α, basic FGF (bFGF), VEGF, and PDGF. These growth factors enable wound healing by causing cell proliferation and synthesis of ECM and inducing angiogenesis. Macrophages play a crucial role in enabling wound healing. Macrophage depletion is known to impair wound closure markedly.4,22
Mast cells
Mast cells are best known for their central role in mediating allergic responses. Beyond that function, it is now known that mast cells are physiologically significant in recognizing pathogens and in regulating immune response.23 Mast cells may instantly release several proinflammatory mediators from intracellular stores. In addition, they are localized in the host–environment interface. These properties make mast cells key players in finetuning immune responses during infection. Recent studies using mast cell activators as effective vaccine adjuvants show the potential of harnessing these cells to confer protective immunity against microbial pathogens.24 Mast cell activation helps initiate the inflammatory phase of wound healing. In response to injury, mast cells at the wound site degranulate within a matter of hours and therefore become histologically silent at the wound tissue. After about 48 hours of injury, mast cells are again seen in the wound tissue and their number increases as healing progresses.25 On one hand, impaired wound healing has been reported in mast cell-deficient mice.26 On the other hand, mast cells have been implicated in skin wound fibrosis.27 With the aid of a wide array of newly formed or preformed mediators released by degranulation, the activated mast cell controls the key events of the healing phases: triggering and modulation of the inflammatory stage, proliferation of connective cellular elements, and final remodeling of the newly formed connective tissue matrix. The importance of the mast cell in regulating healing processes is also demonstrated by the fact that a surplus or deficit of degranulated biological mediators causes impaired repair, with the formation of exuberant granulation tissue (e.g., keloids and hypertrophic scars), delayed closure (dehiscence), and chronicity of the inflammatory stage.28
Macrophages
Macrophages represent the predominant cell type in a healing wound 3–5 days following injury. The primary acute function of wound macrophages, which arrive at an injury site hours later than neutrophils, is to operate as voracious phagocytes cleansing the wound of all matrix and cell debris, including fibrin and apoptotic neutrophils. Macrophages also produce a range of cytokines, growth and angiogenic factors that drive fibroblast proliferation and angiogenesis.4,29–32 In a classic study, Leibovich and Ross33 demonstrated that antimacrophage serum combined with hydrocortisone diminished the accumulation of macrophages in healing skin wounds of adult guinea pigs. Such depletion resulted in impaired disposal of damaged tissue and provisional matrix, compromised fibroblast count, and delayed healing. Today, macrophages have emerged to be a pivotal driver of efficient skin repair.34,35 Macrophages are plastic and heterogeneous cells broadly categorized into two groups: classically activated or type I macrophages, which are proinflammatory effectors, and alternatively activated or type II macrophages.36 In the inflamed tissue, it is unclear whether the type II macrophages that appear during the healing phase originate from newly attracted monocytes or from a switch in the activation state of previously proinflammatory macrophages. The macrophage population first taking part in inflammation may change its phenotype and assume the role to resolve inflammation.37,38 Macrophages from diabetic wounds display dysfunctional inflammatory responses.39 A persistent inflammatory state of diabetic wound macrophages is caused by impairment in the ability of these cells to phagocytose apoptotic cells at the wound site which in turn prevents the switch from M1 to M2 phenotype.39
Resolution of inflammation
Inflammatory responses elicited by injury are only helpful to the healing process if they are timely and transient. Dysregulated inflammation complicates wound healing.39 Complete resolution of an acute inflammatory response is the ideal outcome following an insult. For resolution to ensue, further leukocyte recruitment must be halted and accompanied by removal of leukocytes from inflammatory sites. Resolution of inflammation is executed by a number of key factors. At the wound site successful phagocytosis of dead neutrophils by macrophages is a key factor. Impairment in macrophage function at the wound site derails the resolution of inflammation.39 Lipid mediators, such as the lipoxins, resolvins, protectins, and newly identified maresins, have emerged as a novel genus of potent and stereoselective players that counterregulate excessive acute inflammation and stimulate molecular and cellular events that define resolution.40 Successful resolution paves the path for the healing process to progress towards successful wound closure. Prolonged inflammation may not only compromise wound closure but may also worsen scar outcomes.41,42
Infection
Infection is a common problem in chronic wounds, frequently resulting in nonhealing and significant patient morbidity and mortality.43 Wound infection and the subsequent release of proinflammatory modulators result in pain and delayed healing. The pain, in turn, compromises the immune response to infection.44 All wounds become contaminated by bacteria from the surrounding skin, the local environment, and autologous patient sources. The local environment is particularly relevant for hospitalized patients. Colonization is defined as the presence of proliferating bacteria without a noticeable host response. Colonization of the wound may enhance or impede wound healing, depending upon the bacterial load. Bacterial loads in excess of 105 organisms/gram of tissue are a threat to wound healing, although this threshold may be altered by the status of the host immune system and the number and types of bacterial species present. The concept of critical colonization is controversial and not universally accepted. Critical colonization is characterized by increased bacterial burden or covert infection, and the wound at this stage may enter a nonhealing, chronic inflammatory state. Substantial colonization may not cause the obvious signs of inflammation but will likely affect wound healing with failure to heal or slowing of progression. Signs of critical colonization are atrophy or deterioration of granulation tissue, discoloration of granulation tissue to deep red or gray, increased wound friability, and increased drainage. The transition to infection occurs when bacterial proliferation overcomes the host’s immune response and host injury occurs. Several factors determine transition from colonization to infection: the bioburden itself, the virulence of the organisms, the synergistic action of different bacterial species, and the ability of the host to mount an immune response.43
During the past decade, there has been rapid progress in the understanding of innate immune recognition of microbial components and its critical role in host defense against infection. The early concept of innate immunity was that it nonspecifically recognized microbes; however, the discovery of TLRs (Fig. 14.8) in the mid-1990s showed that pathogen recognition by the innate immune system is instead actually specific, relying on germline-encoded pattern recognition receptors (PRRs) that have evolved to detect components of foreign pathogens, referred to as pathogen-associated molecular patterns (PAMPs).45 TLRs regulate innate and adaptive immune responses and are important modulators of inflammation during wound-healing responses. The finding that there is activation of TLR signaling during tissue damage in several disease situations in the absence of infection suggests that endogenous molecules serve as TLR agonists, although it is unclear whether this response is biologically important for maintenance of homeostasis, such as tissue repair, or whether this recognition is simply accidental. It is noteworthy that microbial infection triggers the production of modified endogenous molecules (such as high-mobility group protein B1, oxidized phospholipids, β-defensin 2, and nucleic acids) that are recognized by TLRs or other cytosolic PRRs. This may suggest that these endogenous molecules, along with PAMPs, act as adjuvants to activate innate immune programs via TLRs and/or other PRRs, and have key roles in facilitating adaptive immunity against infecting microbes.
Chronic wounds have a complex colonizing flora that changes over time. Staphylococcus aureus and coagulase-negative staphylococci are the most commonly isolated organisms. Chronic wounds are colonized by multiple bacterial species and many persist in the wound once they are established. In chronic venous leg ulcers the most common bacteria noted, in order of abundance, were S. aureus, Enterococcus faecalis, Pseudomonas aeruginosa, coagulase-negative staphylococci, Proteus spp., and anaerobic bacteria. Resident (colonizing) bacterial species are commonly present in ulcers. The longer an ulcer remains unhealed, the more likely it will acquire multiple aerobic organisms and a significant anaerobic population. Chronic wounds are commonly complicated by underlying ischemia. Thus, they tend to have a low tissue oxygen level. This facilitates the growth of anaerobes in ischemic wounds. Adequate delivery of oxygen to the wound tissue is vital for optimal healing and resistance to infection.46 Hospitalization, surgical procedures, and prolonged or broad-spectrum antibiotic therapy may predispose patients to colonization or infection, or both, with resistant organisms, including S. aureus (methicillin-resistant S. – MRSA) or vancomycin-resistant enterococci.43 Because inflammatory responses to microbial invasion may be diminished in persons with diabetes, clinical signs of infection are often absent in persons with diabetic foot ulcers when infection is limited to localized tissue.47
Biofilm
The architecture of biofilms is influenced by many factors, including hydrodynamic conditions, concentration of nutrients, bacterial motility, and intercellular communication, as well as exopolysaccharides and proteins.48 Chronic wounds offer attractive conditions for biofilm production because proteins (collagen, fibronectin) and damaged tissues are present, which allow attachment. The biofilm impedes healing of chronic wounds. Most of the chronic wound pathogens, such as MRSA and Pseudomonas spp., are typical biofilm producers. Compared to bacteria in the unattached free-living planktonic form, bacteria that reside within mature biofilms are highly resistant to traditional antibiotic therapies. Bacteria in biofilms grow more slowly, and slower growth may lead to decreased uptake of the drug and other physiologic changes that could impair drug effectiveness.43
Vascularization
Key processes in tissue vascularization are depicted in Figure 14.9. Angiogenic cues are elicited by microenvironmental signals such as hypoxia and are amplified by angiogenic factors such as VEGF expressed by and released from cells at the wound site. VEGF was originally identified as an endothelial cell-specific growth factor-stimulating angiogenesis and vascular permeability. Some family members, VEGF C and D, are specifically involved in lymphangiogenesis. Ligation of these angiogenic factors with their corresponding receptors elicits a multitude of cell-signaling processes that activate microvascular endothelial cells. For example, VEGF and their endothelial tyrosine kinase receptors are central regulators of tissue vascularization. VEGF signaling through VEGFR-2 is the major angiogenic pathway. VEGFR-3 has also been shown to be important for angiogenesis, acting together with VEGF/VEGFR-2 and Dll4/Notch signaling to control angiogenic sprouting.49 The biological significance of other angiogenic factors such as EGF and bFGF is mediated by their corresponding receptors, which also belong to the family of tyrosine kinase receptors EGFR, FGFR-1, FGFR-2, FGFR-3, and FGFR-4.
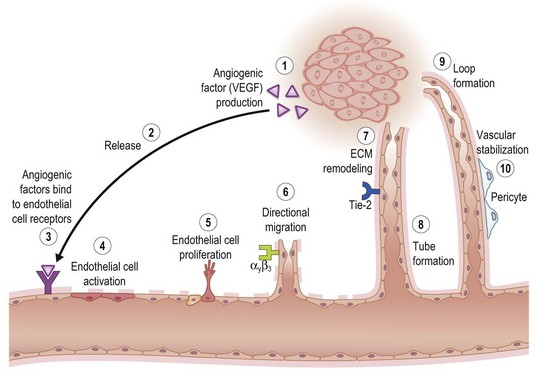
Other tyrosine kinase receptors of outstanding significance in this regard are Tie-1 and Tie-2. Along with the VEGF receptor, these are the only known endothelial cell-specific receptor tyrosine kinases. Tie-2 is induced on the endothelium of neovessels in skin wounds and downregulated as newly formed vessels regress. As an indicator of Tie-2 activation, Tie-2 phosphorylation is detected in skin wounds at all stages of the healing process.50 Activated microvascular endothelial cells respond by proliferating, which is followed by directional migration of these cells. Migration is a complex process in which cells move in a given direction either in response to changes in the extracellular environment or as a consequence of an intrinsic propensity for directional movement. ECM remodeling by proteases promotes cell migration, a critical event in the formation of new vessels. Temporal and spatial regulation of ECM remodeling events allows for local changes in net matrix deposition or degradation, which in turn contributes to control of cell growth, migration, and differentiation during different stages of angiogenesis. Matrix-bound growth factors released by proteases and/or by angiogenic factors promote angiogenesis by enhancing endothelial migration and growth. Matrix molecules promote endothelial cell growth and morphogenesis, and/or stabilize nascent blood vessels. Hence, ECM molecules and ECM remodeling events play a key role in regulating angiogenesis.51
The formation of the capillary-like tubes is specific to endothelial cells and integral to the process of angiogenesis. The basement membrane represents a biologically functional highly specialized ECM on which the basal nonluminal surface of endothelial cells rests. This matrix forms a continuous sleeve around the endothelial cells, and maintains the tube-like structures of the blood vessels. More than 20 years ago Kubota et al. observed that endothelial cells plated on a reconstituted basement membrane matrix, rapidly attached, aligned, and formed capillary-like tubules. The cells did not proliferate. The vessels that are thus formed contain a lumen and tight cell–cell contacts. The cells are polarized with the nuclei basally located towards the basement membrane matrix. Furthermore, the capillary-like structures take up acetylated low-density lipoprotein, which is a marker of differentiation for these cells.52 Angiogenesis not only depends on endothelial cell invasion and proliferation, but also requires pericyte coverage of vascular sprouts for vessel stabilization. These processes are coordinated by VEGF and PDGF through their cognate receptors on endothelial cells and vascular smooth-muscle cells, respectively.53 Structural support to blood vessels is provided to pericytes and vascular smooth-muscle cells. Normal pericytes are embedded within the basement membrane of capillaries, either as solitary cells or a single-cell layer, where they coordinate intercellular signaling with endothelial cells and other components of the blood vessel wall to prevent leakage. In contrast, vascular smooth-muscle cells form single or multiple layers around arteries and veins to mediate vascular tone and contraction. Pericyte coverage is required for the stabilization of immature endothelial tubes.54
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


