Vitiligo: Introduction
|
Epidemiology
The prevalence of vitiligo is reasonably consistent among different populations: ∼0.38% in Caucasians,1 0.34% in Afro-Caribbeans,2 0.46% in Indians,3 though perhaps somewhat less frequent in Han Chinese, 0.093%.4 Vitiligo appears to affect both genders equally, though women are overrepresented among patients seeking clinical care. Vitiligo can develop at any age,5 with a mean age-of-onset in Caucasian patients of about 24 years.6 The most common subtype, generalized vitiligo (GV), is an autoimmune disease that is associated with other autoimmune diseases in about 20%–30% of patients, most frequently autoimmune thyroid disease (Hashimoto’s thyroiditis or Grave’s disease), rheumatoid arthritis, psoriasis, type 1 diabetes (usually adult-onset), pernicious anemia, systemic lupus erythematosus, and Addison’s disease.7
Etiology and Pathogenesis
Vitiligo is a multifactorial, polygenic disorder, with a complex pathogenesis that is not yet well understood.8 Of various theories of disease pathogenesis, the most accepted is that genetic and nongenetic factors interact to influence melanocyte function and survival, eventually leading to autoimmune destruction of melanocytes.7 Other suggested explanations have included defects of melanocyte adhesion,9 neurogenic damage,10 biochemical damage,11 autocytotoxicity,12 and others.
Large-scale epidemiological surveys have shown that most cases of vitiligo occur sporadically, although about 15%–20% of patients have one or more affected first-degree relatives. Typically, familial aggregation of cases exhibits a non-Mendelian pattern suggestive of polygenic, multifactorial inheritance.8 Concordance in monozygotic twins is 23%,6 indicating that both genetic and nongenetic (presumably environmental) factors play major roles in disease pathogenesis.
Almost all studies of vitiligo genetics have focused on GV. Several genes involved in immune function, including loci in the MHC, CTLA4, PTPN22, IL10, MBL2, and NALP1 (NLRP1), have been implicated in susceptibility to GV on the basis of genetic linkage or association studies.7 A recent, very large genome-wide association (GWA) study of European Caucasian GV patients and families identified at least ten different loci that contribute to GV risk.13 Seven of these GV susceptibility loci have also been associated with other autoimmune diseases [(1) HLA class I, (2) HLA class II, (3) PTPN22, (4) LPP, (5) IL2RA, (6) UBASH3A, and (7) C1QTNF6], two loci encode proteins involved in immune function [(1) RERE and (2) GZMB], and another locus, TYR, encodes tyrosinase, a key enzyme of melanin biosynthesis and the major GV autoantigen.
Segmental vitiligo (SV) appears to be genetically distinct from GV. Its generally sporadic occurrence and unilateral distribution have led to the suggestion that it might result from somatic mosaicism for de novo mutations,14,15 perhaps in genes that are critical for melanoblast/melanocyte development or survival, although this hypothesis remains to be confirmed.
There is compelling biological evidence supporting an autoimmune basis for GV.16 GV is epidemiologically associated with a number of other autoimmune diseases,6,17 both in patients and in their close relatives, indicative of a heritable autoimmune diathesis. Humoral immunity was first implicated by the finding in some cases of circulating antimelanocyte autoantibodies18 that target various melanocyte antigens, including tyrosinase, tyrosinase-related protein-1, dopachrome tautomerase, and others, and that have the capability to kill melanocytes in vitro19 and in vivo.20 Currently, these autoantibodies are thought to reflect secondary humoral responses to melanocyte destruction rather than a primary cause of GV.18 A greater role is attributed to the inflammatory infiltrate sometimes seen at the margins of active GV lesions, composed mainly of cytotoxic T lymphocytes. As these T cells express a type-1 cytokine profile and colocalize with epidermal melanocytes, it has been hypothesized that these cells are actively cytolytic toward remaining melanocytes, via the granzyme/perforin pathway.21
An immune mechanism has also been suggested to underlie chemical leukoderma.22 The so-called “occupational vitiligo” may occur in individuals who encounter large doses of phenolic compounds, usually 4-tertiary butyl phenol (4-TBP) and other phenolic compounds that may be contained in cleaning solutions. Occupational vitiligo usually initially involves the hands and forearms (the site of contact with the inciting agent). At present, it is unclear whether these agents are directly toxic to melanocytes, or whether some individuals might be genetically susceptible to melanocyte injury from aliphatic phenolic derivatives, ultimately resulting in melanocyte death, release of antigenic intracellular proteins, loss of tolerance, and autoimmunity.
An immune mechanism has also been proposed for the vitiligo-like depigmentation, which can appear in the course of IL-2 immunotherapy-based treatments for cutaneous melanoma, possibly via the stimulatory effect of IL-2 on T cell growth and activation.23–25 Some melanoma-associated antigens (e.g., MART-1, gp100, and tyrosinase) are expressed on normal melanocytes, suggesting that the occurrence of treatment-related vitiligo-like depigmentation in melanoma may involve cross-reaction of some antimelanoma immune responses with normal melanocytes.
Biochemical Hypothesis
There is some evidence that vitiligo is a disease of the entire epidermis, possibly involving biochemical abnormalities of both melanocytes and keratinocytes.11 The specific morphological and functional abnormalities observed in vitiligo melanocytes and keratinocytes are thought to have a genetic background.11 Ultrastructural abnormalities of keratinocytes from perilesional vitiligo skin have been related to impaired mitochondrial activity,26 and are thought to affect the production of specific melanocyte growth factors and cytokines that regulate melanocyte survival.11 An essential biochemical finding is elevated levels of H2O2 in affected regions of epidermis,27 that may be caused in part by reduced enzymatic antioxidant capacity of keratinocytes and melanocytes.11,19 A defective antioxidant defense may confer melanocytes an increased susceptibility both to immunologic cytotoxicity and to cytotoxicity induced by reactive oxygen species.19
Clinical Features
The principal clinical manifestation of vitiligo is the appearance of acquired milk-white macules with fairly homogeneous depigmentation and well-defined borders. On the basis of the polymorphic distribution, extension, and number of white patches, vitiligo is classified into generalized (vulgaris, acrofacial, mixed), universalis, and localized (focal, segmental, and mucosal) types.28 Vitiligo is also classified as segmental and nonsegmental types, on the basis of distinctive clinical features and natural histories.29 According to this classification, non-SV includes all cases not classified as segmental, including localized, generalized, and acrofacial.
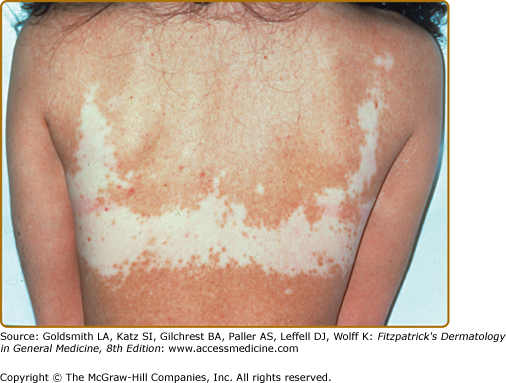
- Vitiligo vulgaris—multiple scattered lesions distributed in a more or less symmetrical pattern; the most common presentation of GV (Fig. 74-1).
- Acrofacial vitiligo—affects the distal end of fingers and facial orifices in a circumferential pattern; a subtype of GV (Fig. 74-2).
- Mixed vitiligo—combination of acrofacial and vulgaris, or segmental and acrofacial types.
- Vitiligo universalis—complete or nearly complete depigmentation of the whole body (Fig. 74-3); the most severe form of GV.
- Focal vitiligo—characterized by the presence of one/few macule(s) in one area but not distributed in a segmental pattern (Fig. 74-4); considered a precursor form of GV.
- Mucosal vitiligo—a term reserved for depigmentation of mucous membrane alone.
- SV—characterized by macules having unilateral dermatomal distribution that do not cross the midline (Fig. 74-5). It generally affects young children and typically remains localized, the depigmented lesions persisting unchanged for many years.30 The occurrence of concomitant other autoimmune diseases is uncommon, compared with GV.29,31,32
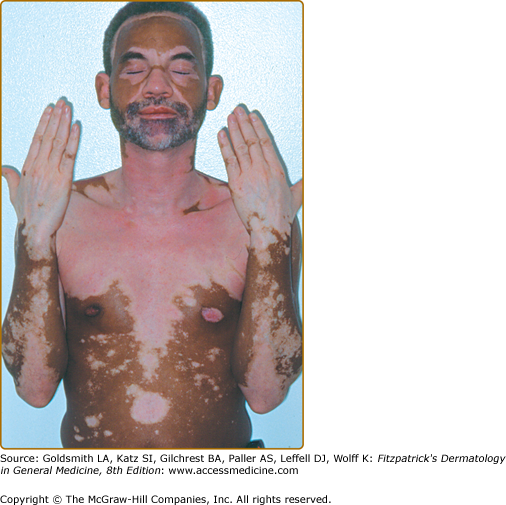
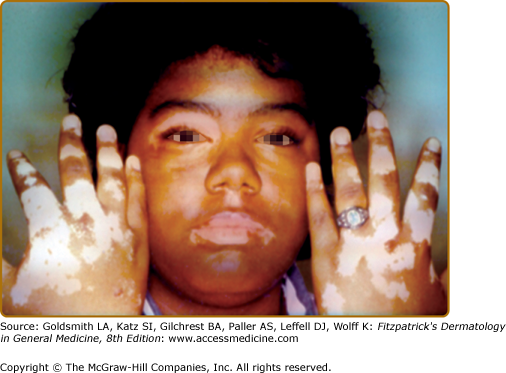
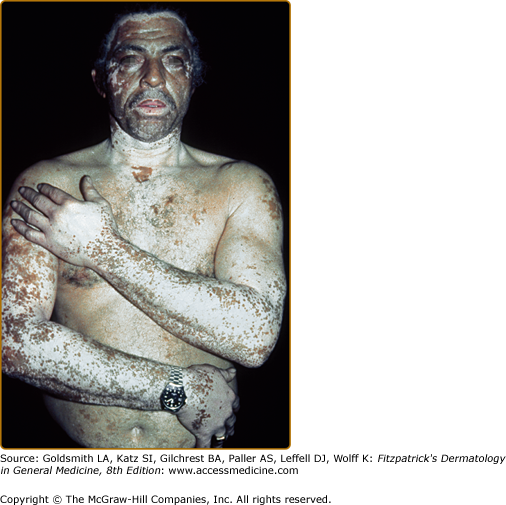

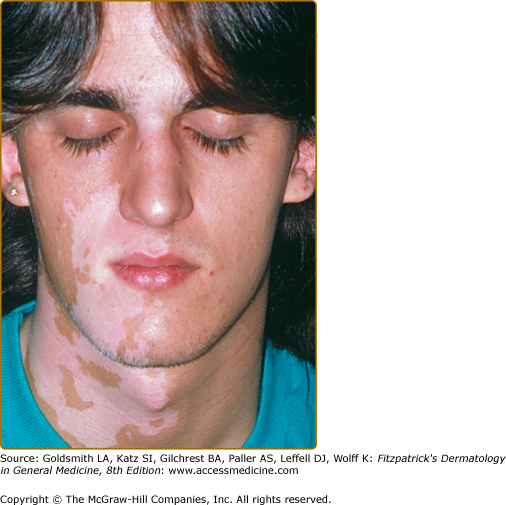
Vitiligo often demonstrates a predilection for sun-exposed regions, body folds, and periorificial areas, although any part of the body can be affected. Various precipitating factors have been suggested, including physical trauma to the skin, sunburn, psychological stress, inflammation, pregnancy, contraceptives, vitamin deficiency, and many others. However, at this time no specific environmental triggers have been proven.
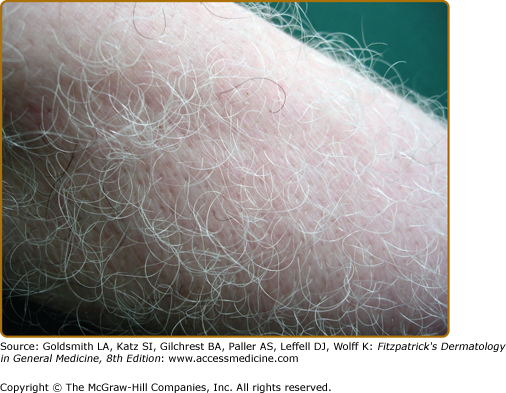
Leukotrichia (depigmentation of hair within vitiligo macules (Fig. 74-7), can be quite variable (10%–60%), and is considered to indicate destruction of the melanocyte reservoir within the hair follicle, therefore, predicting a poor therapeutic response.28 Premature graying of the hair has been described in up to 37% of patients with vitiligo,28 although poor definition and quantitation of this feature makes this supposed clinical association uncertain.
Specific Rare Clinical Phenotypes
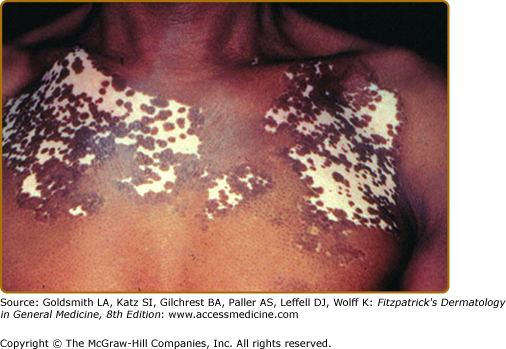
- Trichrome vitiligo is characterized by the presence of patches of intermediate hue (hypopigmentation) between the normal skin and the completely depigmented skin.
- Quadrichrome vitiligo is characterized by the presence of a fourth color (dark brown) at sites of perifollicular repigmentation. It is more often encountered in patients with darker skin phototypes.
- Pentachrome vitiligo—the occurrence of five shades of color: (1) white, (2) tan, (3) medium brown, (4) dark brown, and (5) black.
- Confetti vitiligo or vitiligo ponctué—tiny punctate-like depigmented macules on a hyperpigmented macule or on normal skin.
- Red vitiligo—the depigmented lesions have a raised erythematous border.
- Blue vitiligo—a blue–gray appearance of the skin, which corresponds histologically with the absence of epidermal melanocytes and presence of numerous dermal melanophages.
Related Physical Findings
GV has been often associated with a variety of other conditions, principally autoimmune diseases.33 The most prevalent associated autoimmune disease is autoimmune thyroid dysfunction, either hypothyroidism (Hashimoto’s thyroiditis) or hyperthyroidism (Grave’s disease). Other autoimmune diseases such as rheumatoid arthritis, psoriasis, type 1 diabetes mellitus (usually adult-onset), pernicious anemia, systemic lupus erythematosus, and Addison’s disease also occur with increased frequency in patients with GV.6,17 Most of these disorders, including GV, can occur in various combinations and constitute components of the APECED (APS1) and Schmidt (APS2) multiple autoimmune disease syndromes.
Vitiligo can also be part of the Vogt–Koyanagi–Harada (VKH) syndrome, a multiorgan disorder that affects pigmented structures, such as the eye, inner ear, meninges, and skin.34 Several clinical and experimental studies have pointed to a role of cell-mediated immunity in VKH, particularly involving CD4+ T cells and Th1 cytokines.35,36
Another very rare multiorgan disorder, Alezzandrini syndrome, associates facial skin depigmentation, poliosis, deafness, and unilateral tapetoretinal degeneration of the eye.37 However, many investigators now believe that VKH and Alezzandrini’s syndrome are merely different clinical expressions of the same fundamental disease.38
Skin depigmentation may occur in melanoma patients in three different clinical contexts39: partial depigmentation (“regression”) of the tumor, leukoderma acquisitum centrifugum around the tumor, and vitiligo-like depigmentation, occurring at distant sites. Vitiligo-like depigmentation is thought to be a marker of the patient developing immunity against melanoma cells and to be an indicator of favorable prognosis, especially in advanced stages.25
The most common form of leukoderma acquisitum centrifugum appears around pigmented nevi (called halo nevi), and often progress to spontaneous disappearance of the nevus,39 presumably via lymphocyte-mediated destruction of nevus cells.
The diagnosis of vitiligo is established principally on clinical grounds, which may include distribution and extent of lesions, and natural history of disease.40
Given the association between vitiligo and other autoimmune diseases, several screening laboratory tests are helpful, including T4 and thyroid-stimulating hormone levels, antinuclear antibodies, and complete blood count. Clinicians should also consider testing for serum antithyroglobulin and antithyroid peroxidase antibodies, particularly when patients have signs and symptoms suggestive of thyroid disease.
A skin biopsy is rarely necessary to confirm the diagnosis of vitiligo. Generally, histology shows an epidermis devoid of melanocytes in lesional areas,41 and sometimes sparse dermal, perivascular, and perifollicular lymphocytic infiltrates at the margins of early vitiligo lesions and active lesions, consistent with cell-mediated immune processes destroying melanocytes in situ.42 Some reports42 have suggested that melanocytes may never be completely absent from the depigmented epidermis and that residual melanocytes maintain the capability of recovering functionality. Further studies are needed to clarify this highly debated issue with obvious therapeutic implications.
Condition | Distinguishing Features |
|---|---|
Inherited hypomelanoses Piebaldism Waardenburg’s syndrome Tuberous sclerosis Ito’s hypomelanosis | Stable and circumscribed white patches (with absence of melanocytes) affecting anterior body and limbs; white forelock; autosomal dominant. White forelock, white skin macules, hypertelorism, deafness, ±Hirschsprung disease; multiple genes-autosomal dominant or recessive. Ash leaf hypopigmented macules, facial/periungual angiofibromas, shagreen patches; autosomal dominant. Linear distribution, unilateral or bilateral pattern of hypopigmented macules; sporadic; chromosomal or genetic mosaicism. |
Infectious disorders Tinea versicolor Secondary syphilis Leprosy (tuberculoid/borderline forms) | Hypopigmented lesions, beginning as reddish macules with fine scales upon scraping and seborrheic distribution. At mycologic examination: hyphae and spores. Depigmented round/oval patches (postinflammatory depigmentation) around the neck (necklace of Venus), trunk, limbs, or depigmented patches with peripheral reticular hyperpigmentation (primary lesion). Serological tests for treponemal infection are positive. Depigmented patches with polymorphic presentation, usually accompanied by localized anesthesia; histology: compact skin granulomas. |
Postinflammatory hypopigmentation Discoid lupus erythematosus, scleroderma, lichen sclerosis et atrophicus, psoriasis | Patients have history of preexisting dermatosis. |
Paramalignant hypomelanoses Mycosis fungoides Cutaneous melanoma Autoimmune reactions to advanced melanoma | Hypomelanotic macules or diffuse depigmentation especially in darker skin phototypes. Flat skin patch, plaque, and tumors. Histology: epidermal infiltrates with mononuclear cells. Halo depigmentation around or within the tumor. Depigmentation at a distance from the tumor; the presence of tumor excludes typical vitiligo. |
Idiopathic disorders Idiopathic guttate hypomelanosis Postinflammatory pigment loss | Hypopigmented well circumscribed macules, sharply defined and small in size. They are slow progressive and nonconfluent. Histology indicates epidermal atrophy and reduction in melanin content. Postburn hypopigmentary lesions, shaped in the form of the burn. Hypopigmenting inflammatory reactions leave ill-defined, poorly circumscribed lesions. The history of preceding eruption/injury excludes vitiligo. |
Toxin-induced depigmentation Drug-induced depigmentation | Vitiligo-like depigmentation generally caused by topical occupational exposure to phenolic–catecholic derivatives, often affecting the hands and forearms. Caused by use of systemic drugs (chloroquine, fluphenazine, physostigmine, imatinib, or topical imiquimod). |
NEVUS DEPIGMENTOSUS | Solitary hypopigmented macule well circumscribed with irregular borders, stable in size, solitary, most often present at birth. |
NEVUS ANEMICUS | Hypochromic pale lesion with well-defined borders and irregular margins; often solitary, located on the trunk. Histology and electron microscopic examination reveal no abnormality in melanocytes or melanization. |
Clinical Course and Prognosis
The clinical course of any given case of GV is unpredictable, but is typically gradually progressive and difficult to control with therapy. Sometimes lesions spread over time, whereas in other cases disease activity stops, persisting in stable status for a long period. Some clinical parameters such as a long duration of the disease, occurrence of Koebner’s phenomenon, leukotrichia, and mucosal involvement have been suggested as indicators of relatively poor prognosis.28
Treatment
The key principle of vitiligo therapy is facilitating repopulation of depigmented patches of the interfollicular epidermis with active melanocytes that are able to migrate, survive to repopulate the depigmented skin, and carry out melanin biosynthesis.45 Repigmentation may occur spontaneously and may also be therapy-induced. Spontaneous repigmentation is unpredictable, often clinically insignificant, and tends to be cosmetically unacceptable.46,47 It occurs in fewer than 50% of patients, most commonly in younger patients and in sun-exposed areas, where natural sunlight may act as an inducing agent.48 In clinical practice, the most frequently encountered pattern of repigmentation is perifollicular (Fig. 74-8), though other patterns, such as marginal, diffuse, or combined also may occur.
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


