Vitiligo
Bhavnit K. Bhatia
Shruti Agrawal
Henry W. Lim
Iltefat H. Hamzavi
BACKGROUND
Vitiligo is an acquired pigmentary disorder consisting of depigmented macules and patches due to a selective loss of melanocytes. This condition affects approximately 0.5% to 2% of the worldwide population, with no gender or racial preference. The prevalence in the United States and Europe is estimated to be 1%.1 Although vitiligo can occur at any age, 50% of people develop the condition before 20 years of age.2
Vitiligo may be stable or progressive. In the same patient, it is possible to have simultaneous improvement of some lesions and progression of others. The condition is unique in that it is oftentimes unpredictable, both in terms of progression and therapeutic response to any given treatment. As such, the condition continues to be a therapeutic challenge. Although many therapies can achieve some degree of repigmentation, complete and sustained response is uncommon and difficult to achieve. In this chapter, we outline an approach to vitiligo using an algorithm that takes into account medical, surgical, and supportive treatment options.
PRESENTATION
Patients present with slowly growing, sharply demarcated white macules or patches, often triggered by inciting events such as trauma, stress, and sunburn. Although vitiligo can appear anywhere on the body, it is commonly present as a periorificial loss of pigment on the face and often involves the digits, flexor wrists, elbows, axillae, nipples, umbilicus, knees, and anogenital regions.1
DIAGNOSIS
Clinical Diagnosis
Diagnosis of vitiligo is usually made clinically by examination of the skin in room light and confirmed by
Wood’s lamp illumination, which differentiates depigmentation from hypopigmentation. Distribution of the depigmented patches is used to categorize the subtype of vitiligo. Full body skin examination is essential as genitalia are often affected, and patients may be embarrassed by or even unaware of genital depigmentation. Biopsy of affected skin can help confirm the diagnosis of vitiligo.2
Wood’s lamp illumination, which differentiates depigmentation from hypopigmentation. Distribution of the depigmented patches is used to categorize the subtype of vitiligo. Full body skin examination is essential as genitalia are often affected, and patients may be embarrassed by or even unaware of genital depigmentation. Biopsy of affected skin can help confirm the diagnosis of vitiligo.2
Histopathology
Histology of vitiligo is characteristic for complete absence of melanocytes in the vitiliginous skin. Usually, immunohistologic staining using HMB45, MART1, Mel-5, and others that highlight melanocytes is required to confirm their absence. In stable lesions, there is minimal to no inflammatory infiltrate, whereas active lesions may have a lichenoid interface dermatitis or perivascular lymphocytic infiltrate.2
Subtypes
Broadly, the condition can be categorized into 2 groups: segmental, which does not cross the midline, or nonsegmental vitiligo.3 The types of nonsegmental vitiligo include vitiligo vulgaris, in which there are widely distributed, often symmetric, patches; acrofacial vitiligo, which involves the face and distal extremities; and vitiligo universalis, in which minimal normally pigmented skin remains.
PATHOGENESIS
Vitiligo is caused by a complex interaction between genetic, environmental, and immunological parameters that together make individuals vulnerable to developing the condition.4 Although studies have shown that there is a genetic basis for the development of vitiligo, this does not function in a simple Mendelian pattern. Rather, the genetic component is multifactorial and polygenic.5,6 For example, twin studies have shown that the concordance for generalized vitiligo in monozygotic twins is 23%,7 which provides strong evidence for a genetic component while showing that nongenetic factors play a very important role as well. Studies have shown that up to 20% of patients report a relative with vitiligo,1 and the risk in Caucasian, Indo-Pakistani, and Hispanic first-degree relatives is 7.1%, 6.1%, and 4.8%, respectively.7
A number of genes and susceptibility loci have been identified based on population-based studies. In recent years, over 50 genes have been identified. The large majority of implicated genes encode immunomodulatory proteins, as well as melanocyte components that regulate pigmentary variation, including autoimmune antigens.8,9,10 Many newly identified genes play a role in immune regulation, apoptosis regulation, and melanocyte function and encode proteins that interact both physically and functionally.10 Many of these susceptibility loci are also associated with other autoimmune conditions, providing further evidence for the genetic association of vitiligo with these other disorders.5,10 Up to approximately one-third of patients with vitiligo have increased frequencies of comorbidities, including thyroid disease, pernicious anemia, systemic lupus erythematosus, alopecia areata, rheumatoid arthritis, inflammatory bowel disease, diabetes mellitus, and myasthenia gravis, as compared with the general population.1,5,11
Vitiligo is a multifactorial disorder related to both genetic and environmental factors.8 The selective loss of melanocytes results in the clinically apparent white patches of vitiligo. Patients often identify and attribute vitiligo onset to illness, physical injury, sun exposure, specific medications, pregnancy, and emotional stressors.2 There are major theories on the pathogenesis of vitiligo, including autoimmune, oxidative stress, neural-based, and defective melanocyte adhesion theories. The theory taking hold is convergence theory, in which an oxidative event in melanocytes triggers the immune processes that result in the development of vitiligo.1,12,13
One of the most widely accepted theories of vitiligo is the autoimmune theory. The autoimmune theory of vitiligo describes the destruction of melanocytes by autoantibodies against melanocyte antigens or by cytotoxic T cells.1 Studies have shown increased serum levels of CD8+ T cells in patients with vitiligo compared with healthy controls, and the levels of melanocyte-specific CD8+ lymphocyte levels correlate with disease activity.14,15 Likewise, a decreased ratio of both helper T cells and suppressor T cells has been found in the serum of patients with vitiligo.16 There is also evidence of antimelanocyte antibodies in patients with vitiligo, and levels of antibodies may correlate with disease activity, extent of involvement, and comorbidity with other autoimmune disorders.1 However, what is not yet known is whether this immune-mediated process is primary to the
development of vitiligo or secondary to another process. Studies have also examined the role of cytokines in vitiligo and have found that there is an increase in tumor necrosis factor-α, interferon-γ (IFN-γ), interleukin (IL)-10, and IL-17 in the serum or vitiliginous skin as compared with nonlesional skin.8,9,14,15 The central role of the IFN-γ/CXCL10 pathway in vitiligo pathogenesis is increasingly emphasized. A recent series of studies have shown the central role of the IFN-γ/STAT/CXCL10 pathway in the progression and maintenance of depigmentation. CXCL10, an IFN-γ-induced chemokine, was elevated in skin and serum, and CXCR3, its chemokine receptor, was found to be expressed on pathogenic T cells.17,18,19 The association of vitiligo with other autoimmune disorders further supports an autoimmune etiology.
development of vitiligo or secondary to another process. Studies have also examined the role of cytokines in vitiligo and have found that there is an increase in tumor necrosis factor-α, interferon-γ (IFN-γ), interleukin (IL)-10, and IL-17 in the serum or vitiliginous skin as compared with nonlesional skin.8,9,14,15 The central role of the IFN-γ/CXCL10 pathway in vitiligo pathogenesis is increasingly emphasized. A recent series of studies have shown the central role of the IFN-γ/STAT/CXCL10 pathway in the progression and maintenance of depigmentation. CXCL10, an IFN-γ-induced chemokine, was elevated in skin and serum, and CXCR3, its chemokine receptor, was found to be expressed on pathogenic T cells.17,18,19 The association of vitiligo with other autoimmune disorders further supports an autoimmune etiology.
Individuals with vitiligo have also been reported to have compromised antioxidant responses. For example, oxidant stress levels are thought to be responsible when individuals develop vitiligo after sunburns or exposure to phenolic chemicals.20 Some studies have shown increased levels of reactive oxygen species, including superoxide dismutase, in affected vs nonaffected skin.21 This may affect melanocytes in a number of ways, including effects on signal transduction pathways and disruption of the oxidation-reduction balance in the endoplasmic reticulum of affected skin. Disruption of this balance results in the accumulation of misfolded proteins, which activates the unfolded protein response and results in cessation of normal protein synthesis, upregulation of heat-shock proteins, and even apoptosis.20
Recently, more research has pointed toward a convergence of oxidative stress and autoimmune processes resulting in melanocyte loss.4 One proposed pathway for this is via inducible heat-shock protein 70 (HSP70), postulating that stress causes melanocytes to release HSP70, which subsequently triggers or enhances the autoimmune response in vitiligo.13,22 In one study, skin biopsies comparing lesional and nonlesional skin of patients and age-matched controls demonstrated a correlation between HSP70 expression and disease activity.22
TREATMENT
Management of patients with vitiligo can be broadly categorized into supportive treatment, repigmentation, and depigmentation23 (Algorithm 2.1.1). For the purposes of this text, the medical, ultraviolet/laser, and surgical approaches will be represented with these subcategories indicated accordingly. Repigmentation therapy is appropriate for patients with less than 80% body surface area involvement. Repigmentation can be further subdivided into 3 broad categories of therapy: medical, phototherapy, and surgical. For patients who choose repigmentation, there is consensus developing that early treatment prevents progression of disease.24 In cases in which depigmentation is extensive and involves greater than 80% of a patient’s body surface area, a discussion with the patient regarding pursuing complete depigmentation of normally pigmented skin is warranted.
Medical
Medical treatment of vitiligo consists of supportive care, including camouflage, and topical and systemic therapies.
Supportive
Supportive treatment for vitiligo includes emotional and psychological therapy and cosmetic camouflage options.
Psychological Therapy. Supportive treatment options should be offered to all patients and are not mutually exclusive to medical and surgical therapies. Numerous studies conducted in diverse patient populations have demonstrated the potentially devastating psychosocial consequences of vitiligo. Patients commonly face stigma and experience self-consciousness, fear, worry, lack of self-esteem, and social isolation.25,26,27 This is particularly important given that over 50% of individuals affected are under the age of 20 years, which is a critical period of personal and professional development.28 Studies have shown that these effects may be more pronounced in women, pediatric patients, and patients with Fitzpatrick skin types IV to VI.27,29,30,31 A large study with a broad cohort of patients demonstrated that vitiligo had a significant effect on social and emotional functioning, particularly for women, those with skin types IV to VI, those with generalized vitiligo on the chest, and those who had undergone treatment in the past. Greater effects on quality of life have also been seen with involvement of exposed sites, particularly the hands and face, regardless of total body surface area involvement.32
Studies have also identified that patients with vitiligo are at increased risk of adjustment disorder, anxiety, and
depression.33 Referral to and comanagement with providers in psychology or psychiatry may be warranted. Furthermore, many geographically based support groups exist and should be offered to patients and families.1 The psychosocial effects of vitiligo must be evaluated and managed based on each individual patient’s values, social context, and treatment goals.
depression.33 Referral to and comanagement with providers in psychology or psychiatry may be warranted. Furthermore, many geographically based support groups exist and should be offered to patients and families.1 The psychosocial effects of vitiligo must be evaluated and managed based on each individual patient’s values, social context, and treatment goals.
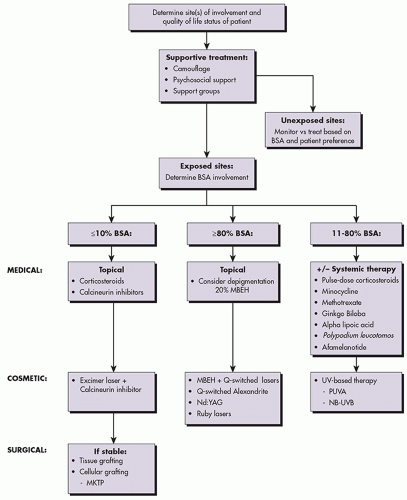 ALGORITHM 2.1.1 Treatment algorithm for vitiligo. BSA, body surface area; MBEH, monobenzyl ether of hydroquinone; MKTP, melanocyte keratinocyte transplantation procedure; NB-UVB, narrowband UVB; PUVA, psoralen with UVA. |
Camouflage. Camouflage is a treatment adjunct that should be offered to all patients expressing concern about the physical appearance of lesions and is integral to the treatment algorithm for patients who report a significant quality-of-life impact. Target populations include young patients and those with involvement of exposed sites, particularly the hands, neck, and face. Before and after studies in both adult and pediatric populations demonstrate a significant improvement of quality of life domains, including clothing choice, embarrassment, self-consciousness, and social interactions.32,34,35,36
Broadly, camouflage can be either temporary or permanent. Permanent methods consist of cosmetic tattoos of various pigments; they have been used successfully for lips. For other body sites, it is difficult to maintain the color match over time, and should vitiligo extend around the tattoo site, it would create a rather unacceptable variation in colors, from normal skin tone, to vitiliginous patches, to tattooed area.37,38 Light and dark brown pigments are achieved using iron oxide.38
Temporary camouflage methods are much more commonly utilized and include makeup and self-tanning products. For camouflaging purposes, makeup must be opaque and designed for more long-term use. Well-established options for all skin types include Dermablend (Division of L’Oreal USA, New York, NY, USA), CoverBlend (Neostrata, Princeton, NJ, USA), and Cover FX (Cover FX Skin Care Inc, Toronto, Ontario, Canada). Newer products include Cavilon 3M No Sting Barrier Film (3M, St. Paul, MN, USA), which makes cover-up more water resistant when applied over makeup.38 Self-tanning products containing dihydroxyacetone are other convenient options. They have been proven to be safe, water resistant, and affordable and can last up to 1 week depending on the site.37,38,39
There are novel camouflage products now available. One such product is Microskin (Microskin, Brisbane, Queensland, Australia), a liquid application that forms a waterproof second skin that lasts 1 to 2 days and can be applied with a sponge or airbrush, depending on the size. Product availability is limited and based in New York, and cost is generally greater than makeup, cream-based, and tanning camouflage products.38 Another emerging product is the Zanderm Vitiligo Concealer (Zanderm, New York, NY, USA), which is marketed as user-friendly pen applicator. It comes in 11 different shades suitable for a wide range of skin types IV to VI and is water and sweat resistant. The product is accessible and affordable, with one pen applicator lasting up to 30 to 60 days depending on coverage area and frequency of use. The product needs to be sealed tightly upon storage as it does dry up rapidly.
Topical
For small body surface areas of less than 10%, topical monotherapy in the form of topical corticosteroids or topical calcineurin inhibitors is first line.40 Studies comparing topical corticosteroids with calcineurin inhibitors show that both are effective options.
Topical Repigmentation
Corticosteroids. Topical corticosteroids generally show a superior response, except in face and genital lesions. Still, this response is variable, rarely greater than 75%, and long-term use is often limited by possible adverse effects.41,42 Class I, II, and III corticosteroids are the topicals of choice, such as clobetasol, betamethasone, mometasone, and fluticasone.43,44,45 These are applied twice a day for a period of weeks to months, and generally a minimum of at least 8 weeks. Corticosteroids are noted to have a significantly higher rate of occurrence of adverse effects as compared with calcineurin inhibitors, including atrophy, telangiectasias, acneiform lesions, folliculitis, and striae.43,44,45,46 As such, a drug holiday after 2 months is recommended.47
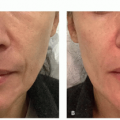
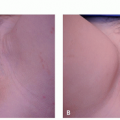
Calcineurin Inhibitors. The calcineurin inhibitors tacrolimus 0.1% ointment and pimecrolimus 1% cream are also commonly used in limited vitiligo, particularly for lesions involving the face, folds, and genitalia (Figure 2.1.1). These are applied twice daily, and duration of therapy is less limited by adverse effects. As compared with topical corticosteroids, they have been found to be safer as monotherapy for facial involvement, and particularly for children.41,43,44,48 The treatment duration in studies is an average of 6 months. The most common adverse effect with calcineurin inhibitors is a burning sensation.41,43,44 Flushing reaction following ingestion of alcohol on calcineurin inhibitor-treated sites, although uncommon, has been well described.49 The utility of
calcineurin inhibitors in combination with phototherapy is now well recognized and further addressed in the Phototherapy section.
calcineurin inhibitors in combination with phototherapy is now well recognized and further addressed in the Phototherapy section.
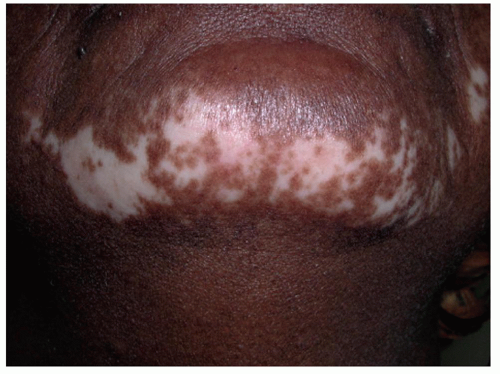 FIGURE 2.1.1 Patient after 6 months of tacrolimus 0.1% ointment applied twice daily to the chin. |
Vitamin D. In some studies, monotherapy with topical vitamin D analogues has been shown to be efficacious in treating vitiligo when applied twice daily at a concentration of 50 µg/g; however, success rates vary and studies show contradictory results.50,51,52 Vitamin D exerts its effect through its nuclear receptor, modulating both pigmentation pathways and T cells involved in melanocyte destruction.53
Combining topical vitamin D treatment with corticosteroids has been shown to have a synergistic effect and more clinical success.42,53,54 In one study, patients who applied betamethasone dipropionate cream 0.05% in the morning and calcipotriol ointment 0.005% in the evening for 3 months were more likely to achieve marked repigmentation and do so at an earlier time point compared with those using either treatment modality individually, although the degree of repigmentation was not statistically significant. Combined treatment may also achieve repigmentation in those who failed to respond to corticosteroids alone.54 Furthermore, by combining corticosteroids and vitamin D analogues, dosages of individual agents may possibly be reduced and decrease the incidence of side effects.42,53
Combination treatment with vitamin D analogues and narrowband ultraviolet B (NB-UVB) is also a safe and successful therapeutic option for vitiligo. Applying topical calcipotriene 0.005% ointment twice daily in conjunction with NB-UVB treatment 3 times weekly resulted in a noticeable enhancement of repigmentation.55 Vitamin D analogues have been shown to potentiate the effect of NB-UVB without adverse effects, resulting in earlier pigmentation, pigmentation of refractory areas, lower UVB dosage, decreased duration of treatment, and decreased cost of treatment.55,56,57 Utilizing natural sunlight may be an efficacious, convenient, and safer option in the pediatric population.58
Pseudocatalase. Oxidative stress is thought to play a role in the pathogenesis of vitiligo. It has been proposed that using topical pseudocatalase in combination with ultraviolet exposure may restore the balance between the oxidant damage and antioxidant enzymatic systems in the skin. An early study demonstrated that applying pseudocatalase and calcium cream twice daily combined with twice weekly broadband UVB treatment resulted in complete repigmentation of the face and hands in 90% of patients.59 However, subsequent studies have since demonstrated contradicting results. A double-blinded, placebo-controlled, randomized trial studied the effects of pseudocatalase cream applied twice daily for 24 weeks combined with NB-UVB treatment 3 times weekly. The results demonstrated that pseudocatalase did not add any benefit compared with treatment with NB-UVB therapy alone.60 Similarly, pseudocatalase mousse applied twice daily combined with twice weekly NB-UVB treatment was not effective in achieving repigmentation.61 In practice, pseudocatalase is no longer used in most centers.
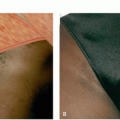
Topical Depigmentation. In cases in which depigmentation exceeds 80% of a patient’s body surface area, complete depigmentation of normally pigmented skin may be considered (Figure 2.1.2). There are various topical methods of depigmentation possible, including monobenzyl ether of hydroquinone (MBEH) 20% to 40%, MBEH in combination with tretinoin for faster results, monomethyl ether of hydroquinone, and 88% phenol, although this may have adverse effects on the liver and kidney in large quantities.62,63
Monobenzyl ether of Hydroquinone. The most commonly employed method is 20% MBEH cream applied topically twice daily; the concentration can be increased to 40% if tolerated. This method has been used dating as far back as 1977, in which 11 of 18 patients achieved dramatic or complete depigmentation after 10 months of use.64 A large retrospective cohort study consisting of 53 patients showed similar results, with 31 patients (58%) achieving complete depigmentation and
an additional 18 (34%) achieving significant repigmentation after about 10 months.65 Of note, 78% of these 49 patients did experience repigmentation after completing treatment, and this was triggered by sun exposure in all but 3 of these patients. Adverse effects were most commonly irritation and irritant dermatitis, which have been noted to be dose dependent.62,63 Maintenance regimens that have been proposed consist of twice weekly treatment with MBEH to prevent repigmentation.
an additional 18 (34%) achieving significant repigmentation after about 10 months.65 Of note, 78% of these 49 patients did experience repigmentation after completing treatment, and this was triggered by sun exposure in all but 3 of these patients. Adverse effects were most commonly irritation and irritant dermatitis, which have been noted to be dose dependent.62,63 Maintenance regimens that have been proposed consist of twice weekly treatment with MBEH to prevent repigmentation.
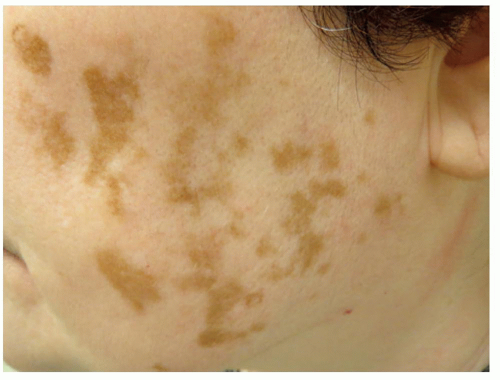 FIGURE 2.1.2 Example of a patient appropriate to consider for depigmentation therapy. |
Systemic
There are a number of systemic therapies, that have more recently been evaluated for use in either slowly or rapidly progressive disease for their ability to halt disease activity or enhance the efficacy of phototherapy (Table 2.1.1). These include pulse corticosteroids, minocycline, methotrexate, afamelanotide, JAK inhibitors, ginkgo biloba, alpha lipoic acid, and Polypodium leucotomos extract. These therapies may be used in combination with topical and NB-UVB therapies.
TABLE 2.1.1 Proposed Effects of Systemic Therapies | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
|
Pulse-Dose Corticosteroids. The clinical utility of systemic corticosteroids is specifically for rapidly progressive or unstable disease.66 Few large-scale studies have evaluated the safety and efficacy of this option. A handful of studies have shown that pulse-dose corticosteroids are effective in halting disease progression, with more variable response in inducing repigmentation. Studies have evaluated dexamethasone 10 or 2.5 mg for 2 consecutive days per week for up to 24 weeks, methylprednisolone 8 mg/kg for 3 consecutive days, and prednisolone taper over 4 months starting at 0.3 mg/kg daily.67,68,69,70,71 In patients with unstable disease, arrest of disease progression was reported to occur in 80% to 90% of patients, whereas repigmentation was highly variable. Few studies have examined the combination therapy of oral corticosteroids plus NB-UVB therapy compared with oral corticosteroids alone; however, they point toward a clinically superior response with combination therapy over oral monotherapy.72 Side effects are uncommon; however, they included weight gain, insomnia, acne, agitation, menstrual disturbance, and hypertrichosis.68,73
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


