Vesiculobullous Disorders
Kristen P. Hook
Jonathan J. Davick
James W. Patterson
INTRODUCTION
Blistering diseases in infants can be a diagnostic challenge that requires consideration of age of onset, pattern of distribution, quality of blistering, and associated extracutaneous signs. They ultimately fall into one of several categories: benign, infectious, genetic, and autoimmune or hypersensitivity reactions. This chapter reviews key clinical signs, along with the pathologic and immunofluorescence findings of many infantile blistering conditions. Correlation among clinical, histologic, immunologic, and serologic data is crucial to establish the correct diagnosis. Genetic testing may be required, as treatments are available to certain mutations, and should be directed by clinical and pathologic data.
Inherited Blistering Diseases
EPIDERMOLYSIS BULLOSA
Epidermolysis bullosa (EB) is a collection of inherited blistering diseases. The main categories include epidermolysis bullosa simplex (EBS), junctional epidermolysis bullosa (JEB), dystrophic epidermolysis bullosa (DEB), and Kindler syndrome (KS).1 Epidermolysis bullosa acquisita (EBA) is an acquired form of EB due to an antibody to collagen 7, and is therefore reviewed in the autoimmune section.
Patients with EB should be phenotypically and genetically categorized. Important phenotypic features include:
The presence of localized or generalized involvement
Mucous membrane involvement
Other cutaneous/appendageal features (tooth enamel defects, hair abnormalities, nail abnormalities, hand/foot deformity)
Extracutaneous features (anemia, esophageal strictures, pyloric atresia)
Skin biopsy for routine histologic analysis may yield minimal diagnostic data. Electron microscopy can provide added information but, because of technical and interpretive limitations, often proves difficult. Immunofluorescence may provide useful information about proteins to target for genetic testing and is quicker than genetic testing. However, with the high sensitivity of available genetic testing, and the high frequency of multiple mutations among affected individuals, genetic testing is recommended for every patient if possible.
When biopsy is used for diagnosis, a newly induced blister should be biopsied for immunofluorescence and electron microscopy. This can be purposefully produced by gently twisting a pencil eraser (or a Q-tip) on a region of skin that is subject to blister formation prior to biopsy. A well-formed blister may not be clinically perceptible, but the microscopic change is usually seen on biopsy.
EB has historically been characterized by eponymous names that indicated a clinical extent of involvement and sometimes diagnostic structural information. These names have been replaced by descriptive terms that state clearly the type, inheritance, and mutation involved. Each EB patient is truly unique. Not only may the specific mutation they carry be unique, but also the way a known mutation manifests phenotypically may vary among individuals, and many
patients carry more than one mutation for varied types of EB that can result in unique phenotypic presentations.
patients carry more than one mutation for varied types of EB that can result in unique phenotypic presentations.
EPIDERMOLYSIS BULLOSA SIMPLEX
Definition and Epidemiology
EBS is a group of inherited blistering disorders characterized by mechanical fragility and blister formation within the epidermis.1 Historically, most cases were associated with mutations in the genes encoding keratin 5 (KRT5) and keratin 14 (KRT14). However, many other culprit genes have now been identified.1,2
EBS is a rare disease, with a prevalence of 6 per 1 million and the incidence of 7.9 per 1 million live births.3 There are two major categories—basal and suprabasal—depending on the level of involvement in the epidermis.1 Further subcategorizations are based on the affected gene/protein and other clinical features.1 The most prevalent subtypes fall into the basal category and include localized EBS (66% of EBS cases), generalized severe EBS (7%), generalized intermediate EBS (6%), EBS with mottled pigmentation (0.4%), and EBS with muscular dystrophy (0.2%).3 The remaining 21% include other rare subtypes, including those affecting suprabasal portions of the epidermis.3
Etiology
EBS is caused by a variety of mutations in genes encoding epidermal structural proteins leading to easy blistering of the skin.1 KRT5 and KRT14 were the first proteins implicated in the pathogenesis of EBS. Many others have since been identified, including plectin, transglutaminase 5, desmoplakin, plakoglobin, plakophilin 1, exophilin, α6β4 integrin, and bullous pemphigoid (BP) antigen-1.1 In one study of 76 patients, KRT5 or KRT14 mutations were identified in 75% of affected patients.4 The most common mode of inheritance is autosomal dominant, with some unusual forms inherited in an autosomal recessive fashion.5
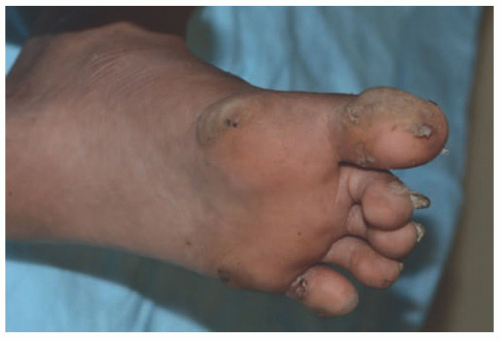 FIGURE 4-1. Epidermolysis bullosa simplex. Note well-formed bullae on the plantar surface. (Courtesy of Kristen Hook, MD.) |
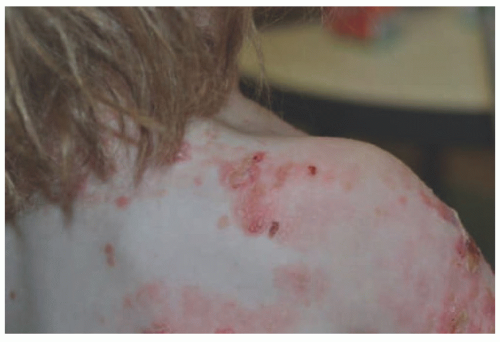 FIGURE 4-2. Generalized epidermolysis bullosa simplex. Flaccid blisters and erosions on an erythematous base. (Courtesy of Kristen Hook, MD.) |
Clinical Presentation
Clinical presentations may include a localized disease of the palms and soles (see Figure 4-1) or generalized disease involving the entire body surface area. Blisters are typically well formed, but rupture easily on the body (see Figure 4-2). Nail dystrophy is a common associated feature (Figure 4-3). Brittle hair or tooth enamel defects may also be associated. Heat and warm weather can increase skin fragility, leading to increased blistering in warm climates or during summer months. Interventions to aid with cooling and drying the skin can be beneficial. Patients typically improve with age, with many young adults experiencing little to no blistering, blistering limited to the foot on warm days. Blisters may be uncomfortable, causing significant pain that precludes standing for long periods of time, which may affect work.
Histologic Findings
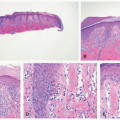
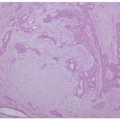
EBS is, by definition, an intraepidermal process, and intraepidermal blister formation is seen histologically (Figure 4-4).6
In the most common basal types of EBS, a split in the epidermis just below the nuclei of the basal layer keratinocytes is most characteristic (Figure 4-4), although superficial layers of the epidermis can become involved with significant trauma.6 Histologic sections for light microscopy may appear to demonstrate a subepidermal blister, as only a small portion of the basal layer keratinocyte cytoplasm may remain attached to the dermis. This is particularly true of older lesions.6 Faint remnants of basal cell keratinocyte cytoplasm are often identified at the base of the blister, and the nucleated half of the split keratinocyte may be found in the blister cavity.6 Blisters are often multiloculated, a feature most commonly seen in early lesions.6 Very early lesions may show only prominent vacuolization (sometimes referred to as “cytolysis”) of the basal layer keratinocyte cytoplasm just below the nucleus6 (Figures 4-4 and 4-5).
In the most common basal types of EBS, a split in the epidermis just below the nuclei of the basal layer keratinocytes is most characteristic (Figure 4-4), although superficial layers of the epidermis can become involved with significant trauma.6 Histologic sections for light microscopy may appear to demonstrate a subepidermal blister, as only a small portion of the basal layer keratinocyte cytoplasm may remain attached to the dermis. This is particularly true of older lesions.6 Faint remnants of basal cell keratinocyte cytoplasm are often identified at the base of the blister, and the nucleated half of the split keratinocyte may be found in the blister cavity.6 Blisters are often multiloculated, a feature most commonly seen in early lesions.6 Very early lesions may show only prominent vacuolization (sometimes referred to as “cytolysis”) of the basal layer keratinocyte cytoplasm just below the nucleus6 (Figures 4-4 and 4-5).
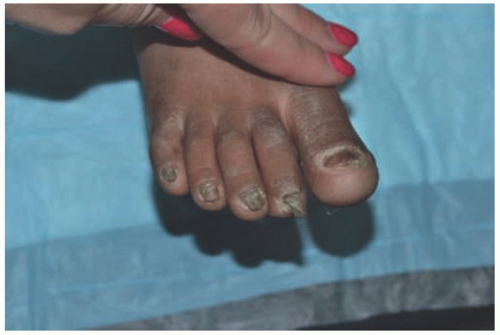 FIGURE 4-3. Nail dystrophy associated with epidermolysis bullosa simplex. (Courtesy of Kristen Hook, MD.) |
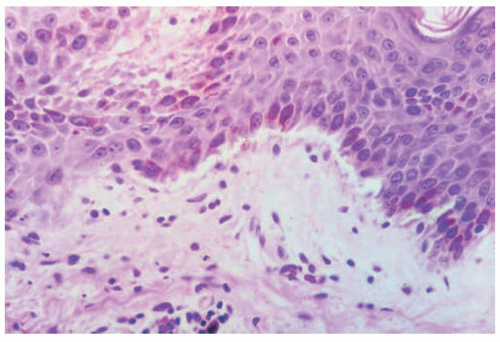 FIGURE 4-4. Epidermolysis bullosa simplex. This is an early lesion; similar changes can be obtained by applying mild trauma to intact skin prior to biopsy. Early clefting can be seen, with some attachment of basal keratinocyte fragments to the base of the separation. |
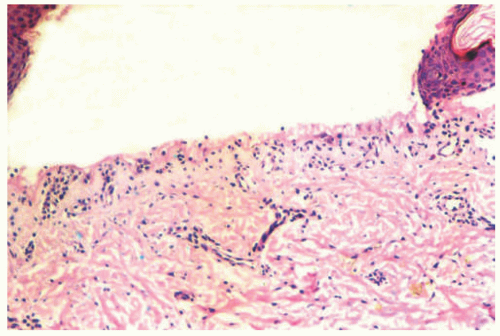 FIGURE 4-5. Epidermolysis bullosa simplex. A fully developed bulla may at first appear to be subepidermal, but the fragments of basal keratinocyte cytoplasm can be seen attached to the dermal side of the separation—indicating the degeneration of the infranuclear portions of basal keratinocytes that characterize this disease. |
As the exact location of the blister may not be apparent on light microscopy alone (intraepidermal vs. subepidermal), immunohistochemical (IHC) stains, direct immunofluorescence (DIF), and electron microscopy (EM) are useful ancillary tests. While EM studies shaped our current understanding of the pathogenesis of EB,7 IHC and DIF methods have largely supplanted ultrastructural examination of routine clinical specimens.
DIF or IHC studies show keratin(s), laminin, and type IV collagen along the floor of the blister. The presence of keratin, together with laminin and type IV collage, proves that small portions of the basal layer keratinocytes are present at the floor of the blister, confirming an intraepidermal process.8
Differential Diagnosis
The differential diagnosis for EBS includes other forms of EB, such as junctional EB or other subepidermal blistering processes. EBS may be distinguished by evidence of an intraepidermal blister. Evidence of cytolysis of the infranuclear portions of basal keratinocytes at the edge of blisters (or sometimes at specimen edges) can help in diagnosis.
CAPSULE SUMMARY
EPIDERMOLYSIS BULLOSA SIMPLEX
EBS is a diverse group of inherited blistering disorders that cause intraepidermal blisters. Inherited mutations in the structural proteins of the skin, the most common of which are KRT5 and KRT14, lead to the clinical manifestations of the EBS diseases. Light microscopy sections show a very low intraepidermal split, most commonly just below the nuclei of the basal layer keratinocytes, although it may appear subepidermal in well-developed lesions. DIF or IHC studies demonstrate keratin(s) and type IV collagen and laminin along the floor of the blister.
JUNCTIONAL EPIDERMOLYSIS BULLOSA
Definition and Epidemiology
JEB is a group of inherited blistering disorders characterized by cleavage in the lamina lucida layer of the epidermis. Mutations in nine different culprit genes have been identified. The prevalence of JEB (all subtypes) is between 0.5 per 1 million and the incidence is 2.68 per 1 million live births.1,3
Etiology
Clinical Presentation
Severe forms of this subtype can be lethal in the first days to months of life. Less severe forms may allow those affected to live for years, but typically with a limitation of some activities and considerable discomfort. Classic signs in the neonatal period include blistering in the diaper area or along the back (Figure 4-6). The blisters may have a “herpetiform” or raised appearance resulting from the granulation tissue. Mucous membranes are commonly involved. Fingernail and hair abnormalities may also be present (Figure 4-7).
Histologic Findings
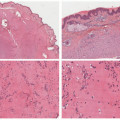
JEB is caused by splitting of the skin at the layer of the lamida lucida, reflected by histologic sections demonstrating a subepidermal blister (Figure 4-8).9 In contrast to EBS lesions, JEB lesions do not show cells or debris within the blister cavity (the so-called cell-free blister is characteristic).9 There is typically no significant associated inflammatory infiltrate.9 Absent or hypoplastic hemidesmosomes can be seen on ultrastructural examination, especially in more severe variants of the disease. IHC or DIF immunomapping identify BP antigen-1 on the roof of the blister, whereas laminin and type IV collagen are identified along the floor.
Differential Diagnosis
The differential diagnosis includes other pediatric blistering disorders discussed in this chapter. The key distinguishing feature is the level of splitting of the skin. Although EBS lesions may appear subepidermal, JEB blister cavities do not typically contain cells or cellular debris as seen in EBS. The differential diagnosis also includes DEB, which can be distinguished by unique immunomapping profiles. Again, correlation with clinical features and molecular genetic studies can aid in the distinction of JEB from other disorders.
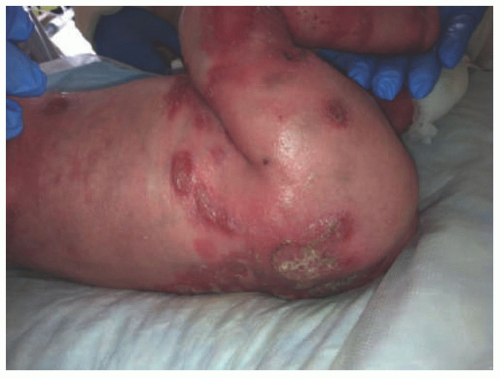 FIGURE 4-6. Junctional epidermolysis bullosa, generalized severe. Note the granulation tissue in wounds. (Courtesy of Kristen Hook, MD.) |
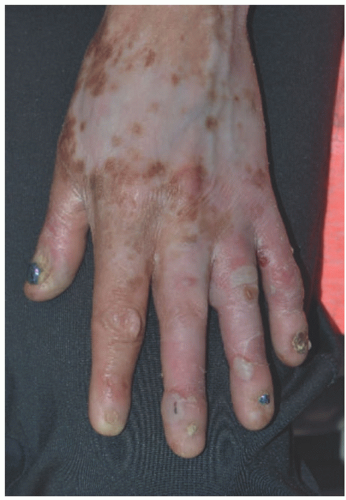 FIGURE 4-7. Junctional epidermolysis bullosa. Note fingernail involvement and dyspigmentation. (Courtesy of Kristen Hook, MD.) |
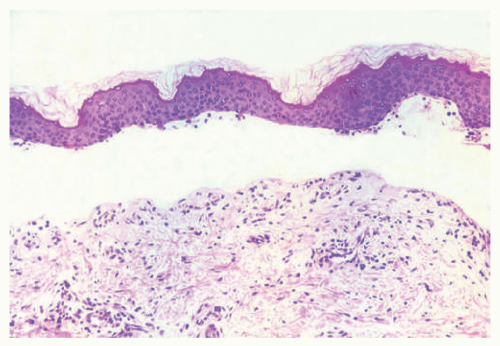 FIGURE 4-8. Junctional epidermolysis bullosa. There is a relatively “clean” subepidermal separation, with a smoothly contoured papillary dermis at the base. A few inflammatory cells are present in this example. Ultrastructural examination may show absent or reduced hypoplastic hemidesmosomes in this condition. |
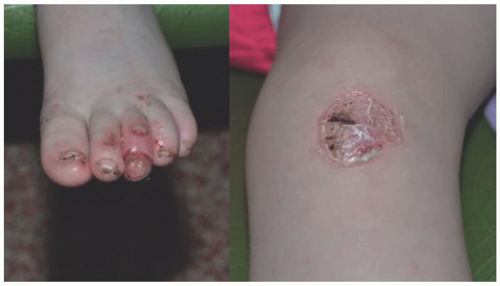 FIGURE 4-9. Dominant dystrophic epidermolysis bullosa. Blistering over knuckles, extensor joints, associated fingernail involvement is common. (Courtesy of Kristen Hook, MD.) |
CAPSULE SUMMARY
JUNCTIONAL EPIDERMOLYSIS BULLOSA
JEB is a group of inherited blistering disorders characterized by blister formation in the lamina lucida of the dermal epidermal junction. Severe forms are lethal in infancy. Histologic sections show a subepidermal blistering without cells in the blister cavity. BP antigen-1 is generally identified on the roof of the blister, whereas laminin and type IV collagen are most often found on the floor of the blister.
DYSTROPHIC EPIDERMOLYSIS BULLOSA
Definition and Epidemiology
DEB is a group of inherited blistering disorders characterized by cleavage in the basement membrane, within the lamina densa.
Etiology
DEB is caused by many described mutations in collagen VII. Cleavage is below the lamina densa in the region of the anchoring fibrils. DEB is inherited in a dominant or recessive pattern.
Clinical Presentation
DDEB is characterized by blisters, atrophic papules, and milia. It most frequently affects the extensor surfaces (interphalangeal joints of the fingers and toes, elbows, knees) (Figures 4-9 and 4-10). Aplasia cutis (absence of the skin) may be a finding at birth (Figure 4-11). Blistering generally improves with age as injury to these areas becomes less frequent. RDEB is characterized by widespread blistering and scarring, joint contractures, mitten deformity of the hands, microstomia, and tooth enamel defects (Figures 4-12, 4-13, 4-14 and 4-15). Generalized RDEB also has many systemic manifestations, including anemia, osteoporosis, and esophageal strictures. Patients with generalized severe subtype have a shortened life span and succumb to aggressive squamous cell carcinomas that form in the areas of frequent blistering. There is broad variation in phenotypic expression, and within these two variants there is much overlap. Patients may present with “mixed” mutations—for example, a collagen 7 mutation that may be associated with DDEB and another that may be associated with RDEB—which creates endless phenotypic heterogeneity.
Histologic Findings
DEB is caused by a split just below the lamida densa, with histologic sections demonstrating a subepidermal blister cavity (Figure 4-16). As with other forms of EB, the lesions
usually lack an inflammatory infiltrate, although chronic inflammation and dermal scarring may accompany the lesions because of continuous skin damage over the course of the disease.10 The numerous subtypes of DEB cannot be distinguished on histologic examination alone, and a specific diagnosis relies on correlation with the clinical and genetic features of the disorder.1
usually lack an inflammatory infiltrate, although chronic inflammation and dermal scarring may accompany the lesions because of continuous skin damage over the course of the disease.10 The numerous subtypes of DEB cannot be distinguished on histologic examination alone, and a specific diagnosis relies on correlation with the clinical and genetic features of the disorder.1
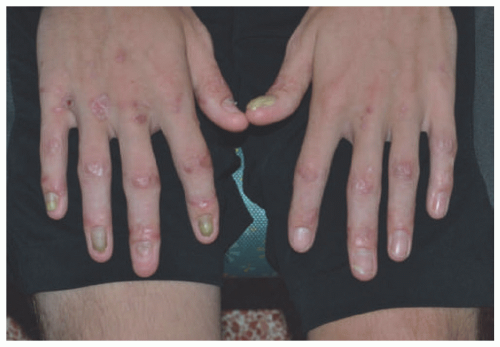 FIGURE 4-10. Dominant dystrophic epidermolysis bullosa. Note milia and scarring over extensor joints and associated nail dystrophy. (Courtesy of Kristen Hook, MD.) |
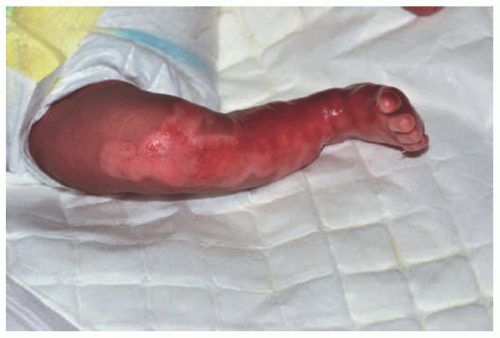 FIGURE 4-11. Aplasia cutis. Congenital absence of the skin can be present in multiple forms of epidermolysis bullosa. (Courtesy of Kristen Hook, MD.) |
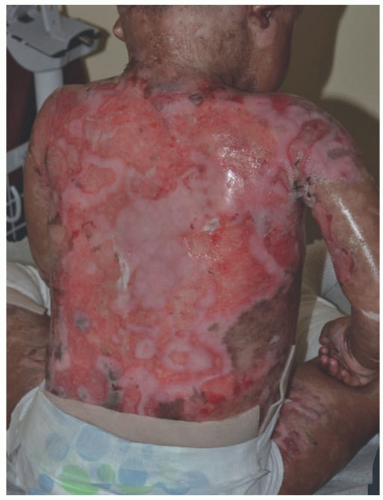 FIGURE 4-12. Recessive dystrophic epidermolysis bullosa, generalized severe subtype. Note generalized erosion, scarring, and dyspigmentation. (Courtesy of Kristen Hook, MD.) |
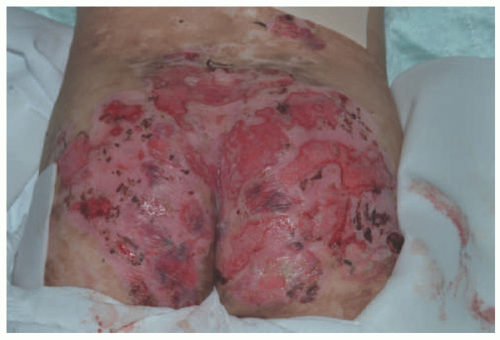 FIGURE 4-13. Recessive dystrophic epidermolysis bullosa, generalized intermediate subtype, involving the buttocks. (Courtesy of Kristen Hook, MD.) |
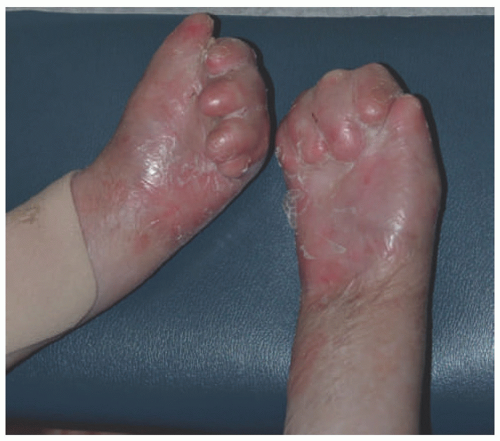 FIGURE 4-14. Recessive dystrophic epidermolysis bullosa, generalized severe subtype with associated mitten deformity. (Courtesy of Kristen Hook, MD.) |
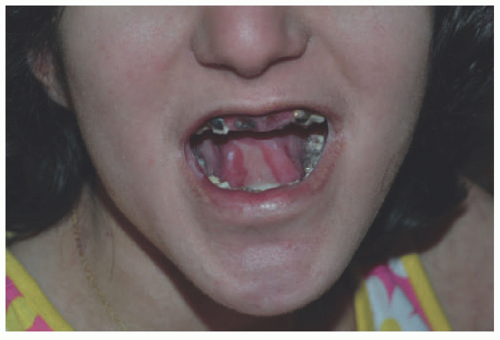 FIGURE 4-15. Recessive dystrophic epidermolysis bullosa. Note absent tongue papillae and enamel defects with associated tooth decay and concomitant microstomia. (Courtesy of Kristen Hook, MD.) |
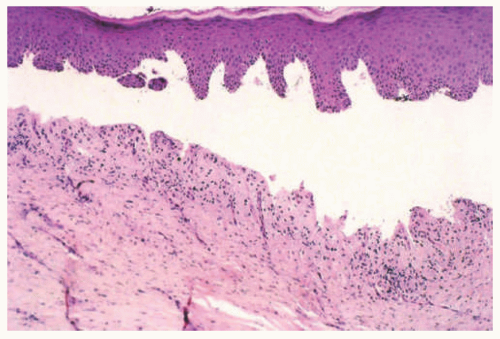 FIGURE 4-16. Dominant dystrophic epidermolysis bullosa. The subepidermal separation and scar are typical features. Some inflammation is present, suggesting that the bulla had been present for some time prior to biopsy. |
Biopsies may be encountered from patients with known DEB to evaluate for squamous cell carcinoma (SCC). SCC is especially common in patients with RDEB (particularly the generalized severe variant), and may occur in adolescents.11,12 Most cases associated with DEB are well-differentiated.11 Despite this, SCCs in this disorder behave aggressively, and the majority of patients with RDEB, generalized severe subtype, die of metastatic SCC within 5 years of diagnosis.11
Milia formation is another well-known histopathologic finding associated with DEB. Milia are small dermal cysts lined by squamous epithelium and containing loose keratin (they are essentially miniaturized epidermal inclusion/epidermoid cysts) (Figure 4-17). In one case report, multiple milia was the presenting skin manifestation of DEB.13 However, any disorder that causes repeated skin damage may result in milia formation, making the finding of milia nonspecific (the lesions of EBA, eg, also frequently show milia). Immunomapping studies show the presence of KRT5 and KRT14, laminins, and type IV collagen along the roof of the blister. On ultrastructural examination, anchoring fibrils are often altered or reduced and may even be absent in severe recessive dystrophic forms of EB.
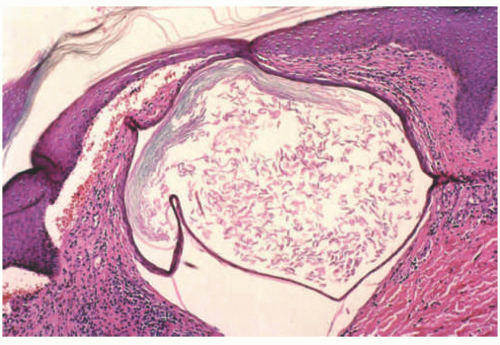 FIGURE 4-17. Recessive dystrophic epidermolysis bullosa. This image shows focal subepidermal separation, dermal scar, and milium formation—a characteristic feature of this form of epidermolysis bullosa. |
Differential Diagnosis
The histologic differential diagnosis of DEB includes other types of EB previously discussed. Milia formation and dermal scarring (features associated with DEB) may provide clues, although neither is specific for the disorder. A lack of cellular debris within the blister cavity can help distinguish DEB from EBS. Immunoreactants mapping to the roof of the blister distinguish DEB from JEB. Once again, correlation with the clinical and genetic features of the disorder is paramount in achieving a definitive diagnosis.
CAPSULE SUMMARY
DYSTROPHIC EPIDERMOLYSIS BULLOSA
DEB is a group of inherited blistering disorders caused by cleavage in the lamina densa of the basement membrane zone as a result of collagen VII mutations. Clinical manifestations include blister formation, milia formation, and scarring and joint contractures in some cases. Patients with generalized severe subtype of RDEB have a shortened life span because of the development of associated aggressive SCCs. Histologic features include subepidermal blister formation, milia, and dermal scarring. DIF and IHC studies will show KRT5/KRT14, laminins, and type IV collagen along the roof of the blister.
KINDLER SYNDROME
Definition and Epidemiology
Etiology
KS results from mutations in the FERMT1 (formerly KIND-1), which encodes fermitin family homolog 1 (FFH1) protein or Kindlin-1, a focal adhesion protein that binds the actin cytoskeleton to the underlying extracellular matrix. The protein regulates cell adhesion and motility by controlling lamellipodia formation in keratinocytes.
Clinical Presentation
KS is characterized by congenital acral blister formation and skin fragility. Photosensitivity, which manifests as progressive poikiloderma, is more prominent during childhood and usually improves with age.15 Diffuse atrophic scarring and
dyspigmentation continue to increase throughout life. Blistering occurs in photosensitive areas and associated atrophy may manifest as pseudosyndactyly (a fusion of the digits secondary to chronic scarring, as seen in the mitten deformity of RDEB). Palmoplantar hyperkeratosis and nail dystrophy are additional features. Mucosal manifestations are varied and can include hemorrhagic mucositis and gingivitis, periodontal disease, premature loss of teeth, and labial leukokeratosis. Severe long-term complications of KS include periodontitis and mucosal strictures (esophageal/laryngeal, anal, urethral, vaginal, and phimosis). Patients may have associated colitis. Patients have an increasing risk of developing SCC in acral skin and oral mucosa, and this risk increases with age.
dyspigmentation continue to increase throughout life. Blistering occurs in photosensitive areas and associated atrophy may manifest as pseudosyndactyly (a fusion of the digits secondary to chronic scarring, as seen in the mitten deformity of RDEB). Palmoplantar hyperkeratosis and nail dystrophy are additional features. Mucosal manifestations are varied and can include hemorrhagic mucositis and gingivitis, periodontal disease, premature loss of teeth, and labial leukokeratosis. Severe long-term complications of KS include periodontitis and mucosal strictures (esophageal/laryngeal, anal, urethral, vaginal, and phimosis). Patients may have associated colitis. Patients have an increasing risk of developing SCC in acral skin and oral mucosa, and this risk increases with age.
Histologic Findings
Reported cases of KS describe hyperkeratosis, epidermal atrophy, vacuolar degeneration of the basal cell layer, telangiectasias (dilation of superficial dermal blood vessels), and pigment incontinence.17,18 Vacuolar degeneration may extend into the areas of subepidermal cleft formation.17,18 Colloid bodies have been described in the papillary dermis.17 Ultrastructurally, multiple planes of cleavage in the dermalepidermal junction may be seen, either in the lamina densa, the lamida lucida, or within the basal layer keratinocytes.18 IHC or DIF studies using type IV or type VII collagen often show reduplication and interruptions of the basement membrane, although this finding is not entirely specific for KS.18
Differential Diagnosis
Because of skin fragility in the neonatal period, there can be clinical phenotypic overlap with DEB and EBS with mottled pigmentation. A diagnosis of KS cannot be made on histopathologic grounds alone as the findings of hyperkeratosis, epidermal atrophy, and vacuolar degeneration of the basement membrane frequently occur in other disorders (especially other poikilodermatous conditions). The finding of reduplication and disruption of the basement membrane on IHC/DIF studies can aid in making the diagnosis but is not diagnostic of KS. Correlation with clinical features, family history, and genetic studies is critical.
CAPSULE SUMMARY
KINDLER SYNDROME
KS is a rare subtype of EB in which patients develop acral blister formation which can progress to severe scarring. Mucosal involvement may be seen. Histologic findings include hyperkeratosis, epidermal atrophy, vacuolar degeneration of the basal cell layer, telangiectasias, and pigment incontinence. IHC or DIF studies show reduplication or interruptions of the basement membrane because of repeated blister formation and healing.
PORPHYRIA AND PSEUDOPORPHYRIA
Definition and Epidemiology
Porphyria is a group of nine inherited disorders caused by altered porphyrin production and characterized by variable photosensitivity and associated systemic signs and symptoms. Variants include acute intermittent porphyria (AIP), hereditary coproporphyria (HCP), variegate porphyria (VP), δ-aminolevulinic acid dehydratase deficiency porphyria (ADP), porphyria cutanea tarda (PCT), hepatoerythropoietic porphyria (HEP), congenital erythropoietic porphyria (CEP), erythropoietic protoporphyria (EPP), and X-linked protoporphyria (XLP). The most common form that presents in the pediatric population is EPP.
Pseudoporphyria demonstrates a photodistributed pattern of blistering but does not result from excessive porphyrin production.
Combined, all forms of porphyria affect roughly 200 000 people in the United States. European studies estimate the prevalence of the most common porphyria, PCT, at 1 in 10 000; the most common acute porphyria, AIP, at approximately 1 in 20 000; and the most common erythropoietic porphyria, EPP, at 1 in 50 000 to 75 000.19 CEP is extremely rare, with prevalence estimates of 1 in 1 000 000 or less. ADP is even more rare, with only isolated case reports in the literature.19 In the pediatric population, the forms most often diagnosed are EPP, AIP, and CEP.
Etiology
Porphyrias are a group of metabolic disorders resulting from abnormal function of the heme biosynthesis pathway and characterized by excessive accumulation and excretion of porphyrins and their precursors. Mutations in the enzymes of the pathway result in overproduction of heme precursors secondary to partial deficiency or, in XLP, increased activity. Each type of porphyria is identified by the enzyme affected and the resultant pattern of porphyrin accumulation. EPP is due to a defect in the enzyme ferrochelatase, which causes accumulation and excretion of protoporphyrin. Liver disease may be an attendant finding in EPP.20
Clinical Presentation
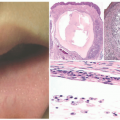
Children may report painful episodes in the sun resulting in severe sunburns. More chronic signs include ephelides, lentigines, and atrophic papules on the dorsal nose and cheeks. The lesions of EPP are usually not blisters, but consist of cobblestoned papules and scars (Figure 4-18). A review of acute porphyrias documented onset between the second and fourth decade of life, with abdominal pain as the most common symptom. Diagnosis is often delayed by more than a decade, and appendectomies and
cholecystectomies are common prior to diagnosis.21 Serologic testing for EPP reveals an elevation of protoporphyrin in the erythrocytes, plasma, bile and feces due to a defect in ferrochelatase enzyme. The diagnosis of EPP is made by demonstrating increased total erythrocyte protoporphyrin and increased percentage of erythrocyte protoporphyrin rather than zinc protoporphyrin. In EPP, metal-free protoporphyrin generally represents more than 85% of the total porphyrins.
cholecystectomies are common prior to diagnosis.21 Serologic testing for EPP reveals an elevation of protoporphyrin in the erythrocytes, plasma, bile and feces due to a defect in ferrochelatase enzyme. The diagnosis of EPP is made by demonstrating increased total erythrocyte protoporphyrin and increased percentage of erythrocyte protoporphyrin rather than zinc protoporphyrin. In EPP, metal-free protoporphyrin generally represents more than 85% of the total porphyrins.
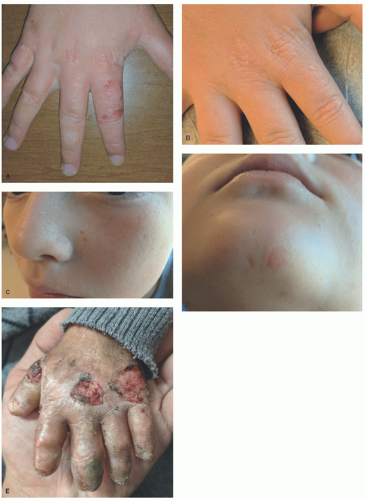 FIGURE 4-18. Porphyria. A, A boy with erythropoietic protoporphyria with a small vesicle and erosions of the dorsal hand during an acute flare. B-D, The same patient after 2 years with subtle atrophic scarring in the sun-exposed areas of the nose, cheek, upper lip, chin, and dorsal hand. E, A child with congenital erythropoietic porphyria. There are erosions on the dorsal hand, and severe, mutilating scarring resulting in shortening of the digits and loss of fingernails. |
Histologic Findings
The different subtypes of porphyrias and pseudoporphyria appear histologically similar. When blister formation is present, histologic sections generally show a subepidermal blister that is free of cells or cellular debris (Figure 4-19). Lesions typically exhibit a small amount of associated chronic inflammation. The key histologic feature in the porphyrias is the presence of thick-walled, hyalinized vessels within the superficial (and sometimes deep) dermis. Periodic acid Schiff (PAS) staining highlights the material surrounding the blood vessels and may also demonstrate PAS-positive globules along the epidermal portion (roof) of the blister (sometimes referred to as “caterpillar bodies”) (Figure 4-20).22,23,24 The lesions may also show sclerosis of the dermis, and festooning (the presence of residual dermal papillae structures pushing into the blister cavity) is often a feature (Figure 4-21).22,23,24 On the basis of ultrastructural studies and antigen mapping, the level of splitting in the blisters is variable, but most often involves the lamida lucida.25 Accordingly, DIF or ICH antigen mapping studies most often demonstrate type IV collagen and laminin along the floor of the blister, and BP-1 antigen along the roof of the blister.25 DIF testing also shows the deposition of C3, IgG, and IgM around vessels in the superficial dermis (sometimes described as “doughnut-like” blood vessels, because of their thickened appearance).26 Indirect immunofluorescence (IIF) is negative in these disorders.
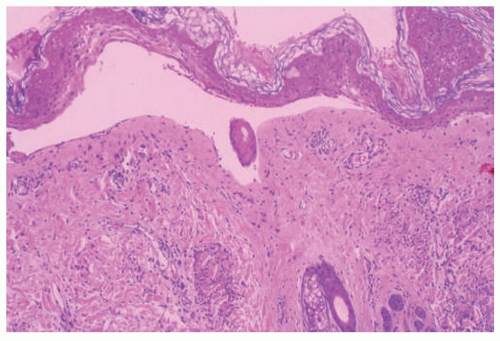 FIGURE 4-19. Porphyria cutanea tarda. There is an infiltrate-poor subepidermal bulla, with some degeneration of the separated epidermis. |
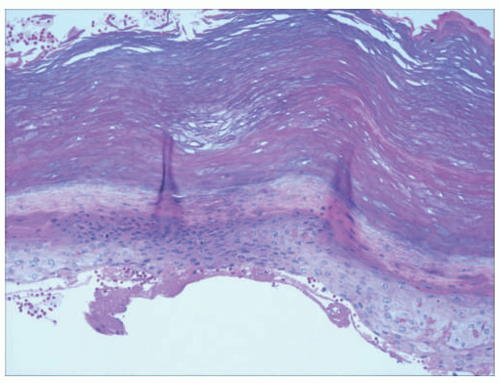 FIGURE 4-20. Porphyria cutanea tarda. The lesion is apparently from an acral location. Globular basement membrane material can be seen at the base of the epidermis; this material forms the so-called caterpillar bodies that are characteristic in this disease. |
Differential Diagnosis
The histologic differential diagnosis includes porphyria and pseudoporphyria. Distinction between these disorders is based on clinical and biochemical laboratory findings. Other disorders with cell-poor blister cavities including EBA, and some forms of EB may also be considered. DIF, IIF, and IHC studies are useful in distinguishing these disorders.
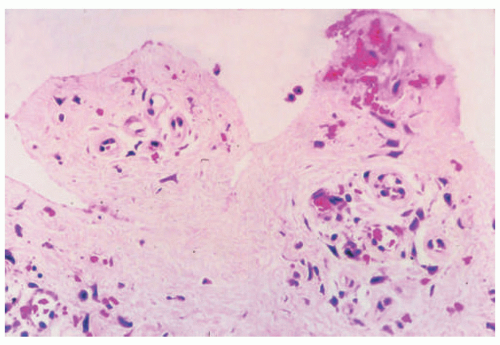 FIGURE 4-21. Porphyria cutanea tarda. This image shows the papillary dermis in another case. Note that the dermal papillae retain their shape (“festooning” of the blister base) and that the capillaries are thick-walled. Inflammation is sparse. |
CAPSULE SUMMARY
PORPHYRIA
The porphyrias are a group of inherited defects in heme synthesis leading to the deposition of porphyrins in the skin and internal organs. Children may present with episodes of severe sunburns with resultant scarring of variable severity, depending on the subtype. Pseudoporphyria is an acquired disorder caused by medications, most commonly nonsteroidal anti-inflammatory drugs (NSAIDS). Histologic sections demonstrate a subepidermal blister with a festooning of the dermal papillae and thickened, hyalinized, “doughnut-shaped” vessels in the papillary dermis that are immunoreactive with antibodies to C3, IgG, and IgM.
Immune-Mediated Blistering Disorders
A review from a pediatric dermatology referral center for a population of 4 million people identified only 23 cases of immunobullous disease in patients less than 18 years of age over a 16-year period.27 Autoimmune causes for blistering result from antibody formation to various proteins in the epidermis, dermis, or attachment proteins (desmosomes, etc). For these disorders, DIF is necessary for diagnosis. Most DIF labs can stain for basic epidermal proteins, but for more complex diagnoses, sendout to specialized labs may be required. IIF is also beneficial (diagnostic in some disorders) and can be used to assess treatment response in many cases.
EPIDERMOLYSIS BULLOSA ACQUISITA
Definition and Epidemiology
EBA results from antibody formation against collagen VII. This is in contrast to DEB, which is attributable to dysfunctional or absent collagen VII.
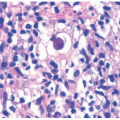
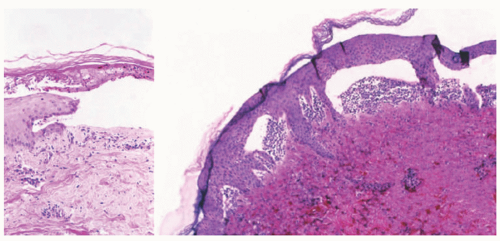 FIGURE 4-22. Epidermolysis bullosa acquisita. The left image shows a relatively infiltrate-poor subepidermal bulla; a few neutrophils can be seen within degenerated portions of the epidermis and in the papillary dermis. The right image shows papillary neutrophilic microabscesses—a feature in some examples of epidermolysis bullosa acquisita that can also be seen in other immunobullous diseases.
Related posts:Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|