Blisters
Vesicles and bullae are the primary lesions in many diseases. Some are of short duration and are quite characteristic, such as those in poison ivy and herpes zoster. In other diseases, such as erythema multiforme and lichen planus, a blister may or may not occur during the course of the disease. Finally, there is a group of disorders in which bullae are present almost continuously during the period of active disease. These autoimmune blistering diseases tend to be chronic, and many are associated with tissue-bound or circulating antibodies. This chapter deals with those disorders.
Autoimmune Blistering Diseases
Autoimmune blistering diseases cause impaired adhesion of the epidermis to the epidermal basement membrane (e.g., the pemphigoid group of disorders [bullous, gestational, and mucous membrane]) or impaired adhesion of epidermal cells to each other (e.g., the pemphigus group of disorders). The autoantibodies target structural proteins that promote cell matrix (e.g., pemphigoid) or cell-to-cell (e.g., pemphigus) adhesion in skin. Autoimmune blistering diseases are characterized by substantial morbidity (pruritus, pain, disfigurement) and, in some instances, mortality (secondary to loss of epidermal barrier function). Bullous pemphigoid (BP) is the most common autoimmune blistering disease. Treatment with systemic immunosuppressives has reduced morbidity and mortality in patients with these diseases.
Major Blistering Diseases
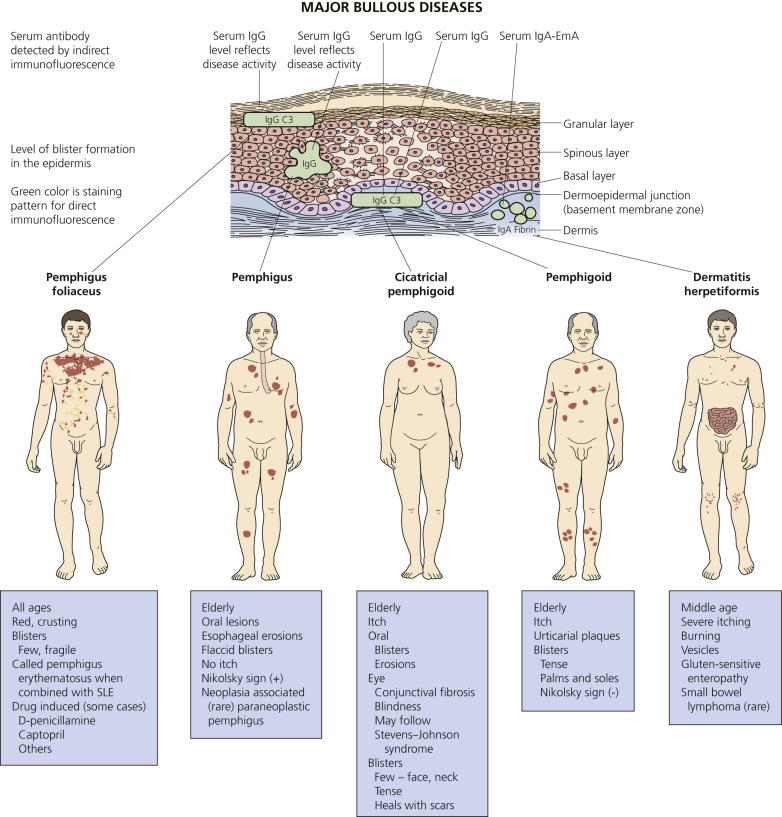
An overview of the major bullous diseases is presented in Fig. 16.1 . The differential diagnosis of all blistering diseases and the anatomy of the epidermis and epidermal basement membrane are shown in Fig. 16.2 .
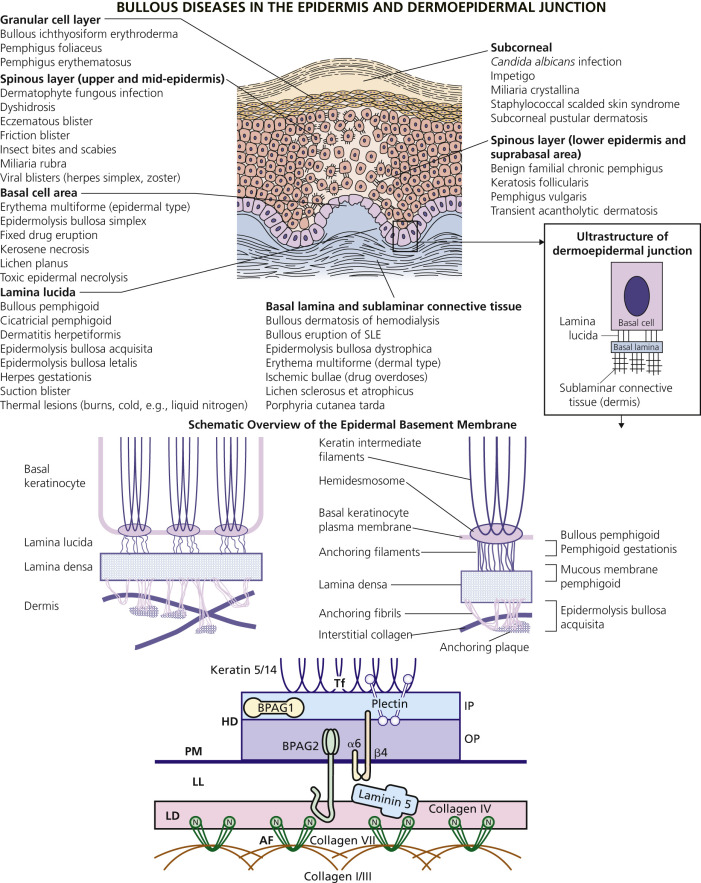
Classification
A blister occurs when fluid accumulates at some level in the skin. The histologic classification of bullous disorders is based on the level in the skin at which that separation occurs (see Figs. 16.1 and 16.2 ). Subcorneal blisters are not commonly seen intact; the very thin roof has little structural integrity and collapses. Intraepidermal blisters have a thicker roof and are more substantial, whereas subepidermal blisters have great structural integrity and can remain intact even when firmly compressed.
Epidermis
Desmosomes contribute to epidermal cell–cell adhesion. Desmosomal proteins (desmoglein 1, desmoglein 3) are the autoantigens of pemphigus foliaceus (PF) and pemphigus vulgaris (PV) ( Table 16.1 ), respectively. Paraneoplastic pemphigus (PNP) is associated with autoantibodies to the desmosomal plaque protein desmoplakin. These diseases have intraepidermal blistering.
| Pemphigus Type | Target Desmosomal Protein |
|---|---|
| Pemphigus vulgaris | Desmoglein 3 (and desmoglein 1) |
| Pemphigus foliaceus | Desmoglein 1 |
| Paraneoplastic pemphigus | Desmoglein 3, desmoplakin 1, desmoplakin 2, BP230, envoplakin, periplakin, others |
| IgA pemphigus | Desmocollin 1 |
The Basement Membrane Zone (See Fig. 16.2 )
The hemidesmosome is a membrane-associated protein complex that extends from the intracellular area of basal keratinocytes to the extracellular area. It links the cytoskeleton of the basal keratinocyte to the dermis. Hemidesmosomes are associated with anchoring filaments, thread-like structures traversing the lamina lucida. Anchoring fibrils, which consist of type VII collagen, extend from the lower portion of the lamina densa to anchoring plaques within the papillary dermis.
Basement Membrane Antigens and Diseases.
Different components of the basement membrane contain the autoantigens of several autoimmune bullous diseases ( Table 16.2 ). Diseases of hemidesmosomes show subepidermal blisters and, by direct immunofluorescence (DIF), linear deposits of C3 or immunoglobulins G and A (IgG and IgA) at the dermoepidermal junction. Bullous pemphigoid is a disease of hemidesmosomes. Two proteins, BP180 and BP230, are the targets of autoantibodies in BP. BP180 is the target of autoantibodies in herpes gestationis (HG), cicatricial pemphigoid, and linear IgA disease. Laminin 5 is an anchoring filament–lamina densa component of hemidesmosomes. Autoantibodies to laminin 5 cause mucous membrane pemphigoid (MMP – cicatricial pemphigoid). Type VII collagen is the major structural component of anchoring fibrils. Type VII collagen is the autoantigen of epidermolysis bullosa acquisita (EBA).
| Bullous Disease | Targeted Molecule |
|---|---|
| Bullous pemphigoid (BP) | BP180, BP230 (hemidesmosome and lamina lucida) |
| Herpes gestationis | BP180, BP230 (hemidesmosome and lamina lucida) |
| Cicatricial pemphigoid | BP180, laminin V (hemidesmosome and lamina lucida) |
| Epidermolysis bullosa acquisita | Type VII collagen (anchoring fibrils) |
| Bullous SLE | Type VII collagen (anchoring fibrils) |
| Linear IgA disease (adults and children) | LAD antigen (BP180) (hemidesmosome and lamina lucida) |
| Dermatitis herpetiformis | Unknown |
Diagnosis of Bullous Disorders
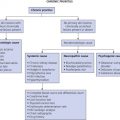
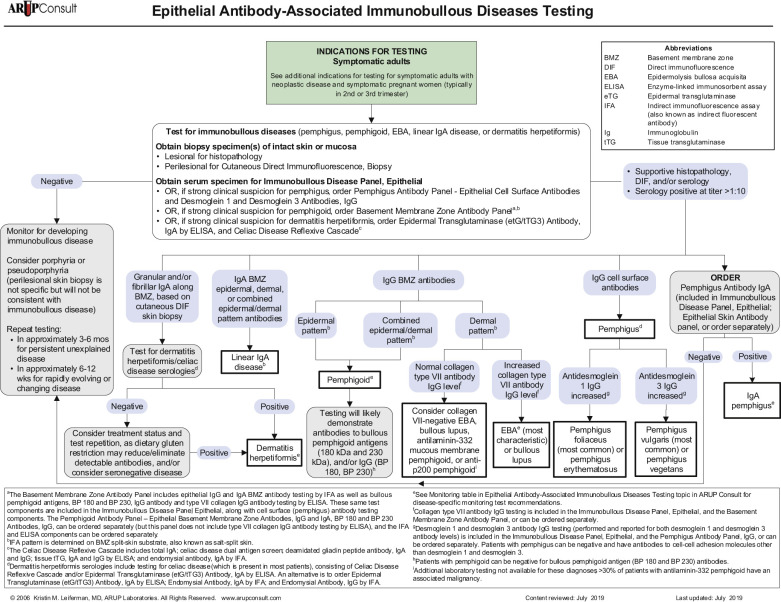
The diagnosis of many chronic bullous disorders can often be made clinically. These diseases have such important implications that the diagnosis should be confirmed by histologic and, in many instances, immunofluorescence studies (see Table 16.3 and Fig. 16.3 ).
| Histopathology | Direct Immunofluorescence | Indirect Immunofluorescence | Diagnosis |
|---|---|---|---|
| Suprabasal |
|
| Pemphigus vulgaris > paraneoplastic pemphigus |
| Subcorneal |
|
|
|
| Subepidermal noninflammatory |
|
|
|
| Subepidermal with eosinophil-rich infiltrate |
|
|
|
| Subepidermal with neutrophil-rich infiltrate |
| Negative on epithelium (+ antiendomysial antibodies)
|
|
Biopsy
For Light Microscopy.
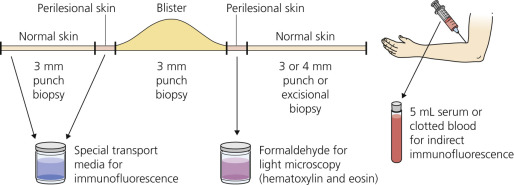
A biopsy specimen must be taken from the proper area to demonstrate the level of blister formation and the nature of the inflammatory infiltrate (see Fig. 16.2 and Table 16.3 ). Small, early vesicles or inflamed skin provides the most diagnostic features. Ruptured or excoriated lesions are of little value and should not be sampled. A small portion of the intact skin should be included in the biopsy specimen. Punch biopsies done through the center of a large blister are of little value.
For Immunofluorescence.
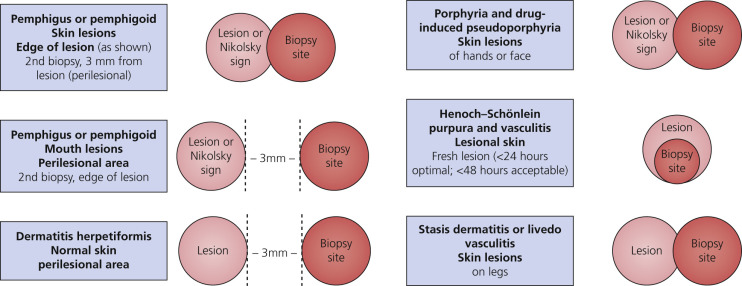
In most cases, the first biopsy specimen should be taken from the edge of a fresh lesion. A second biopsy is often desirable to establish the diagnosis. One should sample skin near the lesion, preferably from a nonedematous, normal, or red area. The best site for obtaining a biopsy specimen is shown in Fig. 16.3 . Fig. 16.4 is a useful algorithm to assist in the diagnosis of immunobullous skin disease.
Level of Blister Formation.
For blisters occurring above the basement membrane zone (BMZ), the level of blister formation can be determined easily with routine studies. Blisters occurring in the dermoepidermal junction (BMZ) area (see Fig. 16.1 ) were once considered subepidermal in location. With the electron microscope, it has been shown that blisters may occur at different levels in that complex area. Electron microscopy is not routinely used; adequate diagnostic information can be obtained from sections stained with hematoxylin and eosin and from immunofluorescence studies.
Immunofluorescence.
Immunofluorescence is a laboratory technique for demonstrating the presence of tissue-bound and circulating antibodies. Most chronic bullous disorders have specific antibodies that either are fixed to some component of skin or are circulating. Many laboratories around the country provide this testing service and supply transport media and mailing containers for tissue specimens.
Direct Immunofluorescence (Skin).
Direct immunofluorescence is designed for demonstration of tissue-bound antibody and complement. Sectioned biopsy specimens are treated with fluorescein-conjugated antisera to human immunoglobulins (IgG, IgA, IgM, IgD, and IgE), C3, and fibrin; they are then examined with a microscope equipped with a special light source.
Indirect Immunofluorescence (Serum).
Indirect immunofluorescence (IIF) is used for demonstration of circulating antibodies directed against certain skin structures. Thin sections of animal squamous epithelium (e.g., monkey esophagus) are first incubated with the patient’s serum. Skin-reacting antibodies in the serum attach to specific components of the animal epithelium. Fluorescein-labeled anti–human IgG antiserum is then added for specific identification of the circulating antibody. The circulating antibody responsible for the IgA deposition has not yet been identified, and IIF is negative.
Dermatitis Herpetiformis and Linear IgA Bullous Dermatosis
Dermatitis herpetiformis (DH) is a rare, chronic, intensely burning, pruritic vesicular skin disease associated in most instances with a subclinical gluten-sensitive enteropathy and IgA deposits in the upper dermis. IgA and antibodies against epidermal transglutaminase 3 (TG3) play an important role in the pathogenesis. DH is most prevalent in patients of northern European decent. The reported prevalence in northern Europe is 1.2 to 75.3 per 100,000 patients. The prevalence in patients in Utah in 1987 was 11.2 per 100,000. Male-to-female ratios range from 1.5 : 1 to 2 : 1. The average age of onset was 41.8 years, with patients having symptoms for an average of 1.6 years before diagnosis. Some studies suggest that DH is more common in males. The incidence of DH appears to be decreasing. Hypothyroidism is the most common autoimmune condition associated with DH. Individuals with DH have an increased risk of B-cell lymphoma. A strong genetic predisposition (human leukocyte antigen [HLA] DQ2 or HLA DQ8 haplotype) to DH exists among affected families. Epidermal transglutaminase appears to be the dominant autoantigen. Dermatitis herpetiformis is rare in children.
Linear IgA bullous dermatosis (LABD) has clinical features similar to those of DH, but has a different histologic and immunofluorescence pattern and there is no associated small bowel disease. Drug-induced LABD is rare but increasing in frequency.
Sulfones produce a dramatic response within hours, but without drugs, some patients have chosen suicide as the only means of relief.
Mayo Clinic Experience
A series of 270 patients were seen at the Mayo Clinic in one study. The mean age at diagnosis was in the fifth decade. The familial incidence was 2.3%. The prevalence of celiac disease (CD) was 12.6% in the patients with DH; diagnosis of CD was made either before the study or during follow-up visits. A high incidence of autoimmune systemic disorders (22.2%) and malignant neoplasms (10.4%) was found. The risk of lymphoproliferative disorders was less than 2%. Histopathologic findings in biopsies of patients with DH were nonspecific in 22% of patients. Endomysial antibodies were present in 63.5% of patients.
Clinical Presentation.
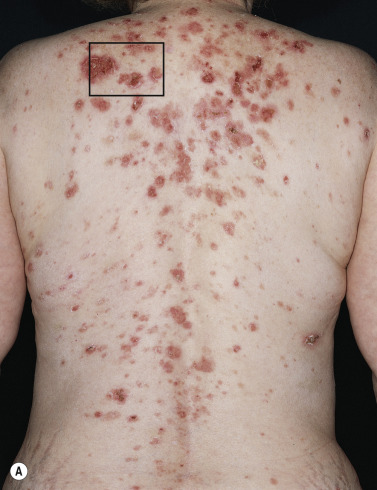
Dermatitis herpetiformis usually begins in the second to fifth decade, but many cases have been reported in children. The disease is rarely seen in blacks or Asians. Dermatitis herpetiformis presents initially with a few itchy papules or vesicles that are a minor annoyance; they may be attributed to bites, scabies, or neurotic excoriation, and they sometimes respond to topical steroids. In time the disease evolves into its classic presentation of intensely burning urticarial papules, vesicles, and, rarely, bullae, either isolated or in groups such as in herpes simplex or herpes zoster (therefore the term herpetiformis) ( Fig. 16.5 ).
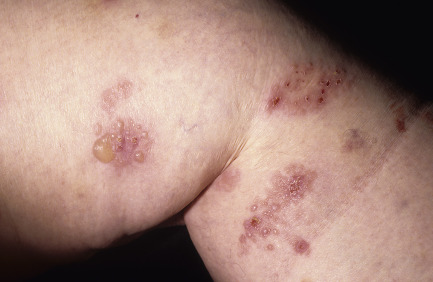
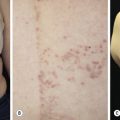
Dermatitis herpetiformis has a classic distribution, involving bilateral extensor surfaces, scalp, and buttocks. The vesicles are symmetrically distributed and appear on the elbows, knees, scalp and nuchal area, shoulders, and buttocks ( Figs. 16.6 to 16.12 ![]() ).
).
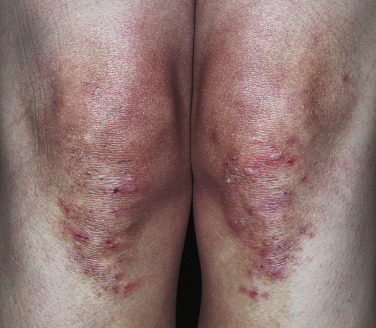
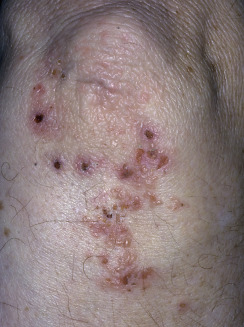
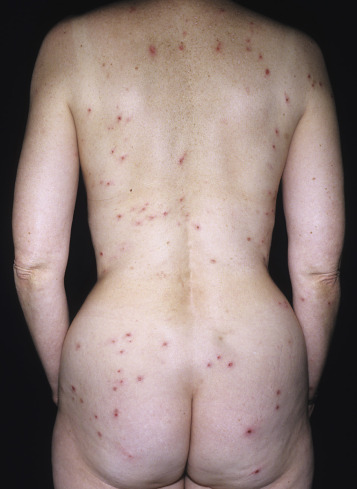
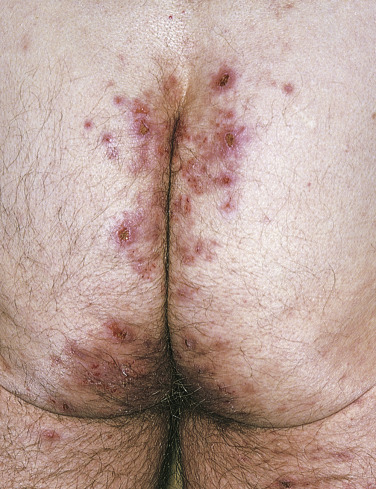
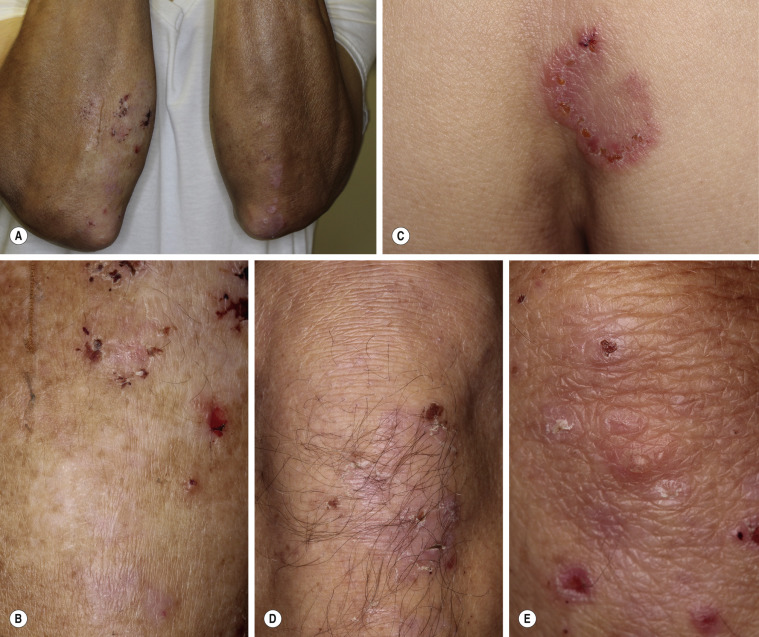
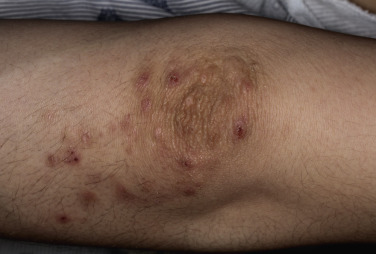
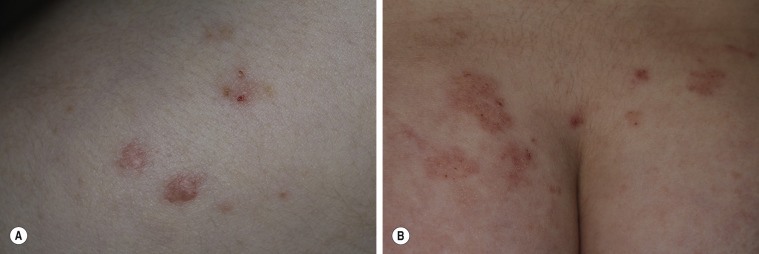
The distribution may be more generalized. Destruction of the vesicles by scratching provides relief but increases the difficulty of locating a primary lesion for biopsy. Intact lesions for biopsy may be found on the back.
The symptoms vary in intensity, but most people complain of severe itching and burning. One should always think of DH when the symptom of burning is volunteered. The symptoms may precede the onset of lesions by hours, and patients can frequently identify the site of a new lesion by the prodromal symptoms. Treatment does not alter the course of the disease. Most patients have symptoms for years, but approximately one third of patients are in permanent remission.
The vesicular–bullous form is confused with bullous erythema multiforme and BP. A strong association between DH and diverse thyroid abnormalities has been reported and most likely represents a grouping of immune-mediated disorders. Hypothyroidism was the most common, occurring in 14% of patients. There were clinical or serologic abnormalities in 50% of patients with DH.
Oral symptoms occur in 63% of patients. Oral dryness and recurrent oral mucosal ulceration are the most commonly reported findings.
Celiac-Type Dental Enamel Defects.
Celiac-type permanent-tooth enamel defects may be found in 53% of patients with DH. The grades of these defects tend to be milder than those described for severe CD. This finding indicates that subclinical gluten-induced enteropathy occurs in early childhood, when the crowns of permanent teeth develop.
Linear IgA Bullous Dermatosis.
Linear IgA bullous dermatosis may present clinically as typical DH, typical BP, or cicatricial pemphigoid or in an atypical morphologic pattern, and it has histologic features similar to those of DH. Drugs such as vancomycin are responsible for some cases. Lesions develop within 24 hours to 15 days after the first dose. The diagnosis of LABD is confirmed by DIF, which shows the presence of linear deposition of IgA at the epidermal BMZ. Some patients demonstrate both IgA and IgG BMZ autoantibodies. There is no gluten-sensitive enteropathy. Some patients have a circulating IgA class anti-BMZ antibody on IIF. Circulating IgA autoantibodies to tissue transglutaminase are detectable in DH but not in linear IgA disease or other subepidermal autoimmune bullous diseases.
Gluten-Sensitive Enteropathy
A gluten-sensitive enteropathy with patchy areas of villous atrophy and mild intestinal wall inflammation is found in the majority of patients with DH. The changes in the small intestine are similar to but less severe than those found in ordinary gluten-sensitive enteropathy; symptoms of malabsorption are rarely encountered. Fewer than 20% of patients have malabsorption of fat, d -xylose, or iron. A significant correlation exists between IgA antiendomysial antibodies (IgA-EmA) and the severity of gluten-induced jejunum damage. Serum IgA-EmA is present in approximately 70% of patients with DH consuming a normal diet. IgA-EmA is positive in 86% of DH patients with subtotal villous atrophy, and 11% of DH patients with partial villous atrophy or mild abnormalities. IgA-EmA antibodies disappear after 1 year of a gluten-free diet (GFD) with the regrowth of jejunal villi. The relationship between IgA-EmA and villous atrophy is a useful diagnostic marker because the enteropathy present in DH is usually without symptoms and therefore difficult to identify.
Lymphoma
Small bowel lymphoma and nonintestinal lymphoma have been reported in patients with DH (B-cell lymphoma) and CD (T-cell lymphoma). Lymphomas occur in patients whose DH has been controlled without a GFD and in those who have been treated with a GFD for less than 5 years. Thus, patients are advised to adhere to a strict GFD for life.
The incidence of lymphoproliferative disorders is less than 2%.
Diagnosis of Dermatitis Herpetiformis
The diagnosis of DH is summarized in Fig. 16.13A .
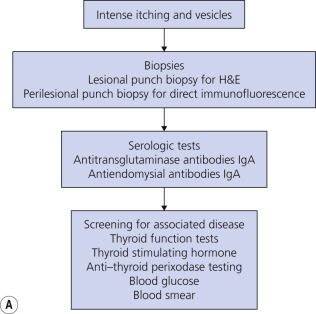
Skin Biopsy.
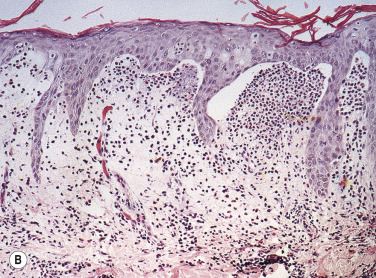
Biopsy an intact vesicle for histopathologic studies. A subepidermal cleft with neutrophils and a few eosinophils is present at the tips of dermal papillae. A mixed perivascular inflammatory infiltrate may be present. Fig. 16.13B illustrates an example of new red papular lesions that have not blistered. Subepidermal clefts of evolving vesicles, and neutrophils and eosinophils in microabscesses within dermal papillae, are demonstrated. Linear IgA bullous dermatosis histologically resembles DH or BP.
Direct Immunofluorescence Studies.
Immunofluorescence studies are necessary for definitive diagnosis. If DH is suspected, take a specimen of normal skin approximately 3 mm from the edge of a lesion. The pathogenic autoantibodies are predominantly of the IgA class. Granular deposition of IgA within the tips of the dermal papillae and along the basement membrane is seen on DIF of perilesional skin. The IgA triggers an inflammatory response that results in a predominantly neutrophilic infiltrate and skin vesiculation. Multiple specimens may be needed to obtain positive findings because of the focal nature of deposits. The immunofluorescence deposits are not altered by dapsone therapy but do slowly resolve on a GFD. Patients with LABD have linear deposits of IgA in the BMZ.
Endomysial Antibodies (IgA).
The finding of IgG-EmA is highly specific for DH or CD. This IIF serum study test is useful for the diagnosis of DH and CD and to monitor adherence to a GFD. Circulating IgA endomysial antibodies are present in 70% to 80% of patients with DH or CD, and in nearly all such patients who have high-grade gluten-sensitive enteropathy and are not adhering to a GFD. The test is negative in normal individuals with DH or in CD patients adhering to a GFD. The titer of IgA-EmA generally correlates with the severity of gluten-sensitive enteropathy. If patients strictly adhere to a GFD, the titer of IgA-EmA should begin to decrease within 6 to 12 months of onset of dietary therapy. A negative result does not exclude the diagnosis of DH or CD. Patients with mild gluten-sensitive enteropathy may have a negative result.
Tissue Transglutaminase, IgA.
Tissue transglutaminase antibody (tTGA) is used as a screening test for DH, in conjunction with the endomysial antibody test, and used for monitoring adherence to a GFD in patients with DH and CD. It is an enzyme-linked immunosorbent assay (ELISA) performed on serum. A total IgA level should be done to rule out selective IgA deficiency, in which case a false-negative test tTGA may be obtained.
Other Studies.
A small bowel biopsy is usually not necessary because of the high sensitivity and specificity of serologic testing. Genetic testing to determine a patient’s HLA haplotype is useful in cases where DH cannot be excluded. See Celiac Disease Diagnostic Testing Algorithms at the Mayo Clinic laboratory website ( https://www.mayocliniclabs.com/it-mmfiles/Celiac_Disease_Diagnostic_Testing_Algorithm.pdf ). Screen for thyroid disease. Check thyroid-stimulating hormone (TSH) and anti–thyroid peroxidase antibody titers.
Measure Blood Glucose Level.
Patients with long-standing, untreated CD, with or without DH, are at a higher risk for developing splenic atrophy. Check a blood smear of DH patients with symptomatic CD to evaluate for the presence of Howell–Jolly bodies (basophilic debris within erythrocytes), which may signify splenic dysfunction. Immunizations against encapsulated organisms (e.g., pneumococcal vaccine) are indicated for those patients.
Trial of Sulfones.
Patients with a classic history of vesicular eruptions may be given a trial of sulfone therapy if they are very uncomfortable. The dramatic relief of symptoms within hours or a few days supports the diagnosis of DH.
Treatment
Dapsone and Sulfones.
These drugs control but do not cure the disease. Dapsone is more effective than sulfapyridine. The mechanism of action is unknown but is possibly explained by lysosomal enzyme stabilization. For adults, the initial dosage of dapsone is 100 to 150 mg given orally once a day. Itching and burning are controlled in 12 to 48 hours, and new lesions gradually stop appearing. The dosage is adjusted to the lowest level that provides acceptable relief; this is usually in the range of 50 to 200 mg/day. Some patients’ symptoms are controlled with 25 mg/day, whereas others may require up to 400 mg/day. Probenecid blocks the renal excretion of dapsone, and rifampin increases the rate of plasma clearance. Dapsone produces dose-related hemolysis, anemia, and methemoglobinemia to a certain extent in all patients. A leukocyte count and hemoglobin determination should be done weekly when possible for the first month, monthly for 6 months, and semiannually thereafter. Methemoglobinemia, although not usually a significant problem, may cause a blue-gray cyanosis. The coadministration of cimetidine, 400 mg by mouth three times a day, is reported to reduce dapsone-dependent methemoglobinemia in DH patients. Patients with glucose-6-phosphate dehydrogenase (G6PD) deficiency may have profound hemolysis during sulfone or sulfapyridine therapy, and those at risk of having the deficiency (blacks, Asians, and those of Mediterranean descent) should have a G6PD level measurement before starting therapy. Dapsone can cause a distal motor neuropathy. This is rare and reversible and requires monitoring by clinical examination. Idiosyncratic side effects, including agranulocytosis, and hepatic function abnormalities occur.
Sulfapyridine and Sulfasalazine.
Sulfapyridine, a short-acting sulfonamide (starting dosage, 500 to 1500 mg/day), can be substituted for dapsone and does not cause neuropathy. It is less effective than dapsone, and patients are usually controlled with 1 to 2 g by mouth daily. Consider sulfapyridine for patients who cannot tolerate dapsone. Sulfapyridine is difficult to obtain in the United States. Sulfasalazine, which is available, is metabolized into 5-aminosalicylic acid (5-ASA) and sulfapyridine. Patients have been reported to respond to 2 to 4 g/day of sulfasalazine. Once sulfasalazine is metabolized in the gut, most of the sulfapyridine is absorbed and excreted in the urine, whereas most of the 5-ASA remains in the gut and is thought to exert a local antiinflammatory effect that makes it useful in the treatment of inflammatory bowel disease.
Adverse Reactions.
Peripheral motor neuropathy may develop during the first few months of dapsone therapy. Generally, high dosages from 200 to 500 mg/day or high cumulative doses in the range of 25 to 600 g have been implicated. Typically, the distal upper and lower extremities, particularly the hand muscles, are involved. Paresthesia and weakness are the most common complaints, and atrophy of interosseous muscles is often found. Patients complain of difficulty with manual tasks and gait disturbance. Footdrop is a common manifestation. Rarely, sensory involvement manifested by paresthesia, diminished pain, and numbness accompanies the motor disorder. Symptoms slowly but invariably improve over months to years when the medication is stopped. Agranulocytosis and aplastic anemia rarely occur but have resulted in death.
Dapsone Hypersensitivity Syndrome.
The dapsone hypersensitivity syndrome (DHS) usually appears 4 weeks or longer after starting the drug. It consists of a mononucleosis-like illness with fever, malaise, and lymphadenopathy. Exanthematous skin eruptions usually resolve within 2 weeks of stopping dapsone. Hypersensitivity hepatitis has been reported as a component of the syndrome. Hypothyroidism may develop 3 months after the onset of DHS. Blood and liver function study results usually become normal within a few months after the patient stops taking dapsone. Prednisone is the preferred treatment. Prednisone is slowly tapered for more than 1 month while the function of affected organs is monitored to minimize recurrences. Dapsone appears to be safe during pregnancy.
Gluten-Free Diet.
Consult a dietitian. Strict adherence to a GFD (avoid foods containing wheat, rye, and barley) for at least 6 months allows most patients to begin a decrease in or possibly a discontinuation of sulfone therapy. The diet has to be followed for many months (often 2 years) before medications can be discontinued. Although intestinal villous architecture improves, symptoms and lesions recur in 1 to 3 weeks if a normal diet is resumed. Current evidence indicates that a GFD needs to be continued indefinitely. Patients found to have a linear IgA immunofluorescence pattern do not have villous atrophy and do not respond to a GFD. Oats can be included in a GFD without deleterious effects to the skin or intestine. Wheat starch–based gluten-free flour products were not harmful in the treatment of CD and DH.
The risk of developing lymphoma may decrease.
Tetracycline and Nicotinamide.
Successful treatment of DH and LABD with tetracycline (500 mg one to three times daily) or minocycline (100 mg twice daily) and nicotinamide (500 mg two or three times daily) is reported. Stopping either nicotinamide or minocycline resulted in a flare-up of the DH. A combination of heparin, tetracycline, and nicotinamide is also reported to be effective.
Bullae in Diabetic Persons
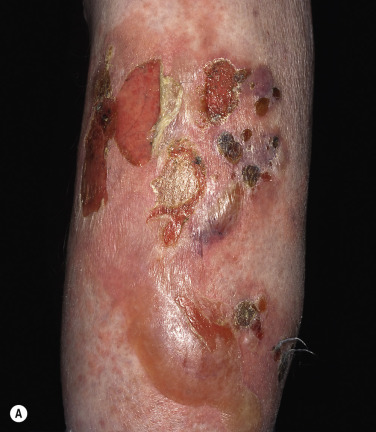
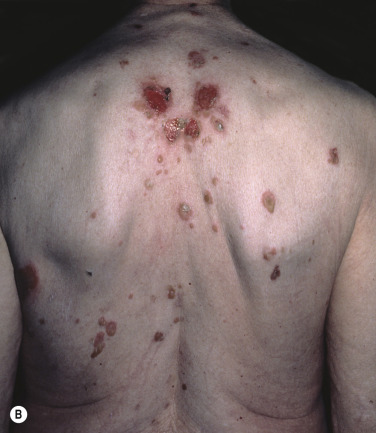
Bullous disease of diabetes (bullosis diabeticorum) is a rare, spontaneous, noninflammatory, blistering condition of unknown etiology occurring in patients with diabetes mellitus. The relationship of the occurrence of diabetic bulla and the degree of metabolic derangement or glycemic control is unknown. It involves approximately 0.5% to 2% of diabetic patients. The male-to-female ratio is 2 : 1 and the age range varies from 17 to 84 years.
Crops of bullae may appear abruptly, usually on the feet and lower legs. The blisters are often large and have an asymmetric shape. Nonacral sites (e.g., the trunk) may also be involved. They usually develop overnight without preceding trauma. There is little pain or discomfort. The bullae arise from a noninflamed base, are usually multiple, and vary in size from 1 cm to several centimeters. Occasionally they are huge, involving the entire dorsum of the foot or a major portion of the lower leg ( Fig. 16.14 ). The bullae are tense and rupture in approximately 1 week, leaving a deep, painless ulcer that forms a firmly adherent crust. Even if not infected, these large ulcers take many weeks to heal. Bullae usually heal spontaneously without scarring. Many patients never have another episode, whereas others have recurrences. It may be complicated with secondary infection. The prognosis is excellent.
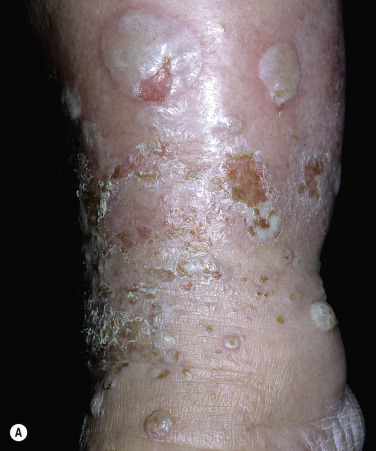
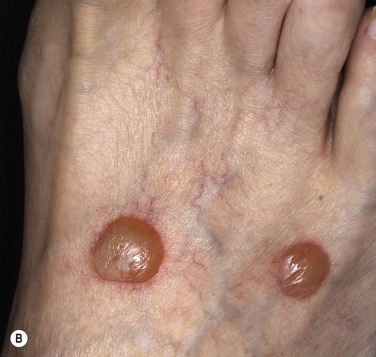
Biopsy shows intraepidermal or subepidermal bulla with minimal inflammation. Direct immunofluorescence reveals no abnormality. The cause is unknown, but is possibly ischemic.
Treatment.
Ulcers may be compressed several times a day with tepid saline solution. The treatment of deeper erosions and ulcers is described in Chapter 3 .
Pemphigus
There are five main variants of pemphigus: PB, PF, pemphigus erythematosus, drug-induced pemphigus, and PNP. Pemphigus (from the Greek pemphix, meaning bubble or blister) is a rare group of autoimmune, intraepidermal blistering diseases involving the skin and mucous membranes. The group includes PV and PF. Both were usually fatal before glucocorticoid therapy was used for their treatment. The difference between the two disorders is the level of the epidermis at which acantholysis (loss of cohesion of epithelium) occurs: the suprabasilar level in PV and the subcorneal level in PF. Other members of the pemphigus group are PNP, which generally occurs in patients with lymphoma, and drug-induced pemphigus, which usually develops after taking penicillamine and other drugs.
Pathophysiology
Pemphigus is the result of the interaction between genetically predisposed individuals and possibly some exogenous factor. The presence of foci of PF (fogo selvagem) in rural South America suggests that the disorder can be triggered in susceptible persons by an environmental agent (probably an unidentified infectious agent), whose antigens mimic those of desmoglein 1 (Dsg1), and that clinical disease evolves in the persons who have the most vigorous immune response to desmoglein 1. Both idiopathic pemphigus and induced pemphigus have the same HLA pattern.
Desmoglein
The structural integrity of the epidermis depends on the sharing of desmosomes by neighboring keratinocytes. Desmosomal connections are broken and re-formed as keratinocytes migrate from the basal layer toward the skin surface. Desmoglein is a cell-to-cell adhesion molecule contained in desmosomes that contributes to the strength of the intercellular desmosomal bridge. There are three isotypes of desmoglein: Dsg1, Dsg2, and Dsg3. Dsg2 is expressed in all desmosome-possessing tissues (see Table 16.1 ); Dsg1 and Dsg3 are restricted to stratified squamous epithelia, where blister formation is found in patients with pemphigus.
Dsg1 and Dsg3 Autoantibodies.
Circulating IgG autoantibodies are directed against the normal desmoglein proteins within the desmosomal structure on the cell surface of keratinocytes. The autoantibodies destroy the adhesion between epidermal cells, allowing fluid to accumulate in the gaps in the epidermis to form blisters. The target molecule of pemphigus autoantibodies is a transmembrane desmosomal component – Dsg3 in PV and Dsg1 in PF.
Patients with mucosal-dominant PV are negative for anti-Dsg1 antibodies but positive for Dsg3 antibodies. Patients with mucocutaneous PV have both anti-Dsg3 and anti-Dsg1 IgG antibodies. Patients with PF have only anti-Dsg1 IgG. Antibodies to other desmosomal cadherins such as anti-desmocollin-3 may be detected in PV and PF and can induce blisters. Ultrastructurally, desmosomes are split, reduced in number and size, and keratin is retracted in pemphigus patients. These structural abnormalities cannot be explained by direct interference of autoantibodies alone, and it is likely that intracellular signaling pathways are altered by the autoantibodies, leading to disruption of desmosomal processing.
Pemphigus Vulgaris
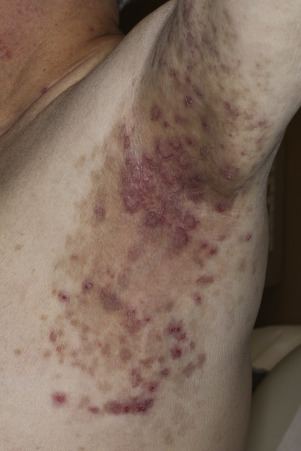
Pemphigus vulgaris is the most common form of pemphigus. Painful oral erosions usually precede the onset of skin blisters by weeks or months ( Figs. 16.15 to 16.17 ![]() ). Involvement of other mucosal surfaces occurs in patients with widespread disease. The soft palate is involved in 80% of cases at initial presentation. Nonpruritic flaccid blisters varying in size from 1 cm to several centimeters appear gradually on normal or erythematous skin and may be localized for a considerable time. The most commonly involved sites are the scalp, face, axillae, and oral cavity. Blisters invariably become generalized if left untreated ( Fig. 16.18 ). The blisters rupture easily because the vesicle roof, which consists of only a thin portion of the upper epidermis, is fragile ( Figs. 16.19 and 16.20 ). Application of pressure to small intact bullae causes the fluid to dissect laterally into the midepidermal areas altered by bound IgG (Nikolsky sign).
). Involvement of other mucosal surfaces occurs in patients with widespread disease. The soft palate is involved in 80% of cases at initial presentation. Nonpruritic flaccid blisters varying in size from 1 cm to several centimeters appear gradually on normal or erythematous skin and may be localized for a considerable time. The most commonly involved sites are the scalp, face, axillae, and oral cavity. Blisters invariably become generalized if left untreated ( Fig. 16.18 ). The blisters rupture easily because the vesicle roof, which consists of only a thin portion of the upper epidermis, is fragile ( Figs. 16.19 and 16.20 ). Application of pressure to small intact bullae causes the fluid to dissect laterally into the midepidermal areas altered by bound IgG (Nikolsky sign).
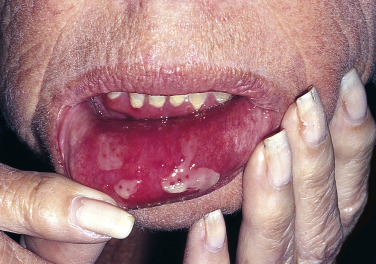
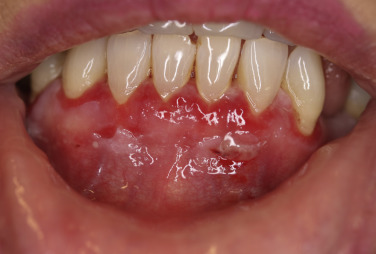
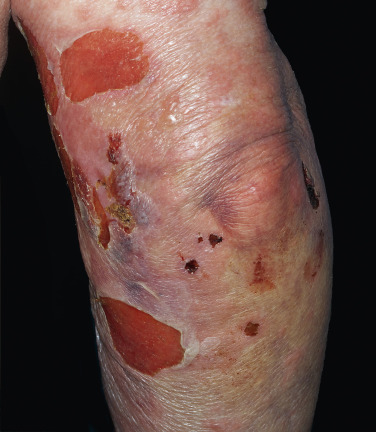
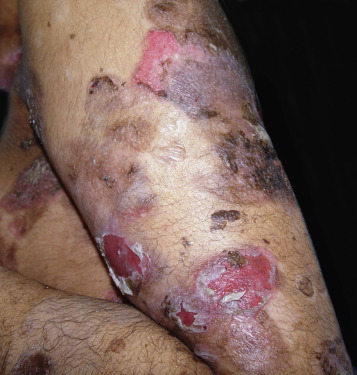
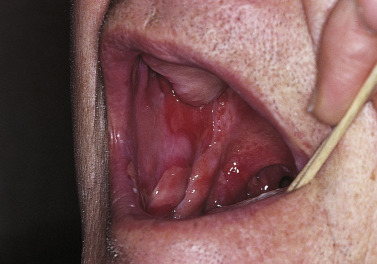
Exposed erosions last for weeks before healing with brown hyperpigmentation, but without scarring. Blisters, erosions, and lines of erythema may appear in the esophageal mucosa. Death formerly occurred in all cases, usually from cutaneous infection, but now occurs in only 10% of cases, usually from complications of steroid therapy. Sunlight exposure is harmful.
Pemphigus vegetans is a variant of PV that presents with large verrucous confluent plaques and pustules localized to flexural areas in the axillae and groin.
Pemphigus Foliaceus, IgA Pemphigus, and Pemphigus Erythematosus
The age of onset varies more widely in PF and pemphigus erythematosus than in PV, and there is no racial prevalence. Oral lesions are rarely present. Pemphigus foliaceus begins gradually on the face ( Figs. 16.21 and 16.22 ) in a “butterfly” distribution or first appears on the scalp, chest, or upper back in a seborrheic distribution.
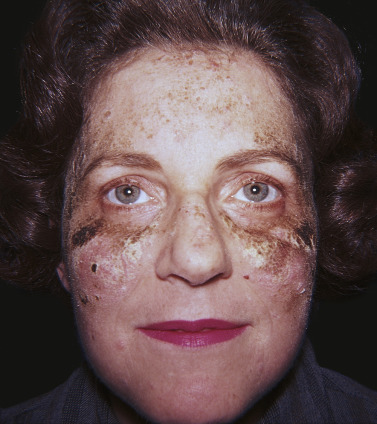
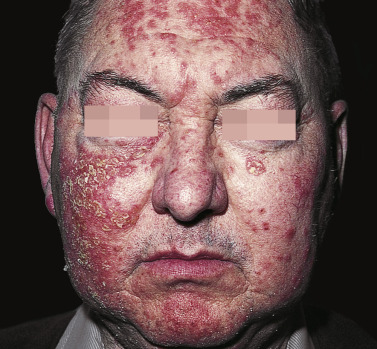
Pemphigus Erythematosus
Pemphigus erythematosus, also known as Senear–Usher syndrome, may actually be a combination of localized PF and systemic lupus erythematosus, because many of these patients have a positive antinuclear antibody. If the eruption becomes more diffuse or generalized, the term PF is used. The disease may last for years and may be fatal if not treated.
Pemphigus Foliaceus
Pemphigus foliaceus (superficial pemphigus) presents with recurrent shallow erosions, erythema, scaling, and crusting. Small flaccid blisters may occur but they are superficial, very fragile, and rupture easily ( Fig. 16.23 ![]() ). Serum leaks from ruptured vesicles and desiccates, forming the localized or broad areas of crust ( Fig. 16.24 ). Intact thin-walled blisters are sometimes seen near the edge of the erosions. The site of blister formation in the horizontal plane of the stratum corneum can be demonstrated in skin biopsy specimens after the upper portion of the epidermis has been dislodged with lateral finger pressure (Nikolsky sign). IgA pemphigus has clinical and histologic similarity to subcorneal pustular dermatosis and PF. IgA antibodies are bound to the epidermal cell surface, and half of the patients have circulating IgA anti–cell surface antibodies.
). Serum leaks from ruptured vesicles and desiccates, forming the localized or broad areas of crust ( Fig. 16.24 ). Intact thin-walled blisters are sometimes seen near the edge of the erosions. The site of blister formation in the horizontal plane of the stratum corneum can be demonstrated in skin biopsy specimens after the upper portion of the epidermis has been dislodged with lateral finger pressure (Nikolsky sign). IgA pemphigus has clinical and histologic similarity to subcorneal pustular dermatosis and PF. IgA antibodies are bound to the epidermal cell surface, and half of the patients have circulating IgA anti–cell surface antibodies.
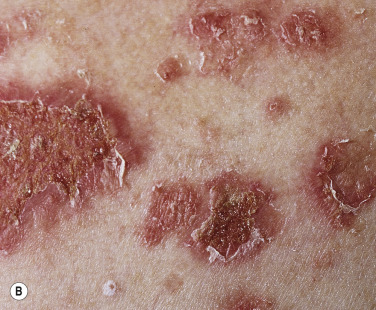
Skin lesions of PF are generally well demarcated and do not extend into large eroded areas as those of PV. Mucous membrane involvement is uncommon. Pemphigus foliaceus may remain localized for years and it has a better prognosis than PV.
Fogo Selvagem
Fogo selvagem (Portuguese for “wild fire”) is an endemic form of PF found in certain rural areas of South America, including Brazil, Colombia, Bolivia, Peru, and Venezuela, as well as in Tunisia. It occurs in jungle areas but the disease disappears when the jungle is cleared. Environmental triggers such as an infectious agent have been proposed to induce fogo selvagem. Fogo selvagem occurs in children and young adults and may affect several family members. This endemic variant is clinically indistinguishable from nonendemic PF.
Diagnosis of Pemphigus
Skin Biopsy for Light Microscopy.
Histologic studies may yield key findings in cases that are negative by DIF. A small, early vesicle or skin adjacent to a blister biopsied with a 3- or 4-mm punch shows an intraepidermal bulla and acantholysis (separation of epidermal cells near the blister following dissolution of the intercellular cement substance). The basal epidermal cells become detached from each other but remain attached to the basement membrane. There is a mild to moderate infiltrate of eosinophils ( Figs. 16.25 to 16.27 ![]() ).
).
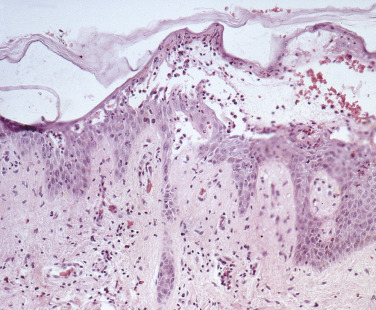
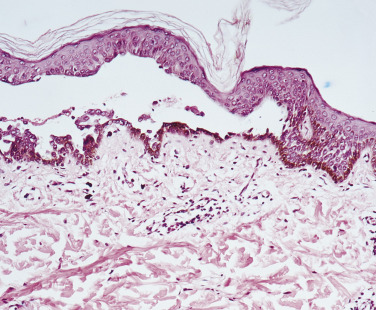
Direct Immunofluorescence of Skin and Mucous Membranes.
If pemphigus is suspected, perform a skin biopsy with approximately the following composition: two thirds normal skin and one third edge of lesion. Two biopsies are recommended. One biopsy specimen should be taken from the edge of a fresh lesion and the second from an adjacent normal area. Two biopsies are especially helpful in evaluation of oral lesions because lesional sites are frequently denuded. Mucosal biopsies should be taken 0.5 cm from a lesion because closer sites may be negative. Perilesional biopsies of skin or mucous membranes usually reveal IgG, strong IgG4, and frequently C3 in the intercellular areas in patients with all clinical variants of pemphigus.
Indirect Immunofluorescence.
Serum IgG antibodies directed against the keratinocyte cell surface can be demonstrated by indirect immunofluorescent staining and are present in all forms of true pemphigus in approximately 75% of patients with active disease. Blood should be withdrawn into a red top tube. A combination of IIF on two substrates (monkey and guinea pig esophagus) and ELISA tests for Dsg1 and Dsg3 affords the greatest sensitivity and highest level of confidence in the diagnosis of pemphigus. ELISA for Dsg1 and Dsg3 distinguishes between PV and PF. Cases that have both skin and mucosal lesions have both Dsg3 and Dsg1 antibodies (see Table 16.1 ).
In some cases, the level of circulating intercellular substance IgG antibody reflects the activity of disease, rising during periods of activity and falling or disappearing during times of remission. Many patients show a poor correlation between the titer of circulating antibody and disease activity. Therefore management of pemphigus should be guided by clinical disease activity rather than by the pemphigus antibody titer. In some cases periodic serum tests to detect changes in titers are helpful in evaluating the clinical course. Serum should be tested every 2 to 3 weeks until remission and every 1 to 6 months thereafter.
Treatment
There are no clear treatment guidelines for PV at this time .
Topical Steroids.
Patients with mild PV or PF responded to clobetasol propionate 0.05% cream applied to mucosal lesions and involved skin twice a day for at least 15 days, followed by progressive tapering.
Corticosteroids and Immunosuppressive Agents.
Systemic glucocorticosteroids are still the mainstay of therapy. Very-high-dose regimens (more than 120 mg/day) provide no benefit over the low-dose regimens with respect to the frequency of relapse or the incidence of complications.
The majority of PV patients present with oral disease at an early and relatively stable stage. These patients may be controlled by starting prednisone 40 mg on alternate days plus 100 mg azathioprine every day until there is complete healing of all lesions. A gradual monthly and later bimonthly decrease of prednisone was followed by the tapering of azathioprine, in a 1-year period. The time required for the epithelialization of lesions varies between 4 and 7 months.
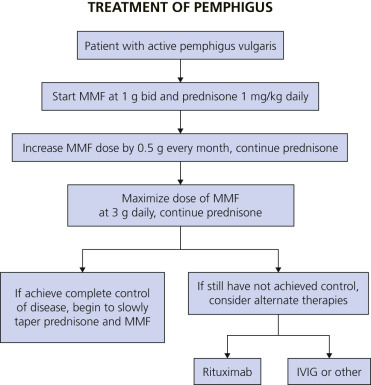
The combination therapy of mycophenolate mofetil (MMF) and prednisone is reported to be an effective treatment regimen to achieve rapid and complete control of PV. For those patients who fail treatment with MMF and prednisone, rituximab is an efficacious alternative therapy. Complete disease control was achieved in 89% of patients using the treatment algorithm in Fig. 16.28 ![]() .
.
Mycophenolate Mofetil.
Average starting dosage is 1 g/day. Adverse effects are gastrointestinal disorders (most common), genitourinary complaints, increased incidence of viral or bacterial infection, and neurologic symptoms. Relative contraindications include lactation, peptic ulcer disease, hepatic or renal disease, and concomitant azathioprine or cholestyramine therapy. Detailed prescribing information is found in Box 8.19 ![]() .
.
Rituximab.
Rituximab is a chimeric murine–human monoclonal antibody that targets the CD20 antigen found on B cells and results in rapid depletion of this cell population. It is indicated for patients with relapsed or refractory, low-grade or follicular, CD20-positive, B-cell non-Hodgkin lymphoma. It has been used for refractory PV, PF, and PNP. The rheumatoid arthritis dosage of rituximab (1000 mg × 2, days 1 and 15) is efficacious and well tolerated in patients with pemphigus. Patients who fail to achieve remission after 1 cycle or patients who relapse seem to benefit from repeated rituximab cycles. Infectious adverse events are a risk with use of this biologic agent. The combination of rituximab and intravenous immunoglobulin (IVIG) is effective in patients with refractory PV. Patients have also been treated with the lymphoma protocol for a single cycle (375 mg/m 2 ) for 4 consecutive weeks. Approximately 80% to 85% of patients will receive rapid clinical response with rituximab, but the majority of patients require ongoing immunosuppressive therapy to maintain clearance, albeit at a lower dose than before rituximab infusions. Rituximab may also be utilized with corticosteroids as a steroid-sparing agent. Recently, rituximab has been FDA-approved for treatment of PV. The FDA-approved dose is two 1000-mg intravenous infusions separated by 2 weeks in combination with a tapering course of glucocorticoids. The maintenance treatment is a 500-mg intravenous infusion at month 12 and every 6 months thereafter or based on disease severity. To treat PV relapse, rituximab is administered as a 1000-mg intravenous infusion on relapse alone with administration of glucocorticoids.
Intravenous Immunoglobulin.
Intravenous immunoglobulin (400 mg/kg/day for 5 days) in a single cycle is an effective and safe treatment for patients with pemphigus who are relatively resistant to systemic steroids.
Adjuvants.
Because of the potential toxicity of systemic corticosteroids, another drug may be initiated long term. The adjuvant therapy (corticosteroid-sparing medication) is initiated with or after starting corticosteroids. Although there are no controlled studies most experts believe that immunosuppressive agents have a steroid-sparing effect. They may decrease the side effects of steroid therapy by allowing the use of lower steroid dosages and lead to increased remission rates. Others disagree and feel that the improved prognosis of pemphigus in recent years is due to the use of lower dosages of corticosteroids, and the improved treatment of corticosteroid complications. The most commonly used agents are cyclophosphamide, and azathioprine and MMF. Methotrexate (MTX) is rarely used because of the reported high incidence of severe infections. One study showed that MMF and azathioprine had similar efficacy, corticosteroid-sparing effects, and safety profiles as adjuvants during treatment of PV and PF.
Cyclophosphamide.
Average starting dosage is 100 mg (1.1 to 2.5 mg/kg) per day. Cyclophosphamide may be the most effective drug but it is toxic. Side effects include bone marrow suppression, hemorrhagic cystitis, bladder fibrosis, reversible alopecia, and an increased risk of bladder carcinoma and lymphoma. Monitor urinalysis and blood cell counts. Encourage oral fluid intake to decrease the risk of bladder fibrosis and hemorrhagic cystitis.
Azathioprine.
Average starting dosage is 100 mg (1.1 to 2.5 mg/kg) per day. Azathioprine causes bone marrow suppression, hepatotoxicity, and an increased risk of malignancy that is lower than that of cyclophosphamide. Monitor blood cell counts and liver function tests. Detailed prescribing information for azathioprine is found in Box 8.20 ![]() .
.
Many other agents have been tried alone or as adjuvants. These include chlorambucil, MMF, dapsone, gold, plasma exchange, extracorporeal photopheresis, and tetracycline 2 g/day.
Approach to Treatment
Therapeutic choices are determined by the patient’s age, degree of disease involvement, rate of disease progression, and subtype of pemphigus.
Prednisone.
Elderly patients with mild to moderate disease can be treated with prednisone, at 40 mg/day, along with cyclophosphamide or azathioprine. Patients with more severe disease may require 60 to 80 mg/day of prednisone. The dosage of prednisone is tapered to a level that controls most disease activity. Attempts are made to use an alternate-day regimen to minimize side effects.
One taper method is to reduce prednisone by 10 mg every week until the daily dose reaches 20 mg. Then the dose is reduced each alternate week until a dose of 20 mg on alternate days is reached. Then the dose reduction is slower until a final dose of 5 mg on alternate days is achieved.
During the prednisone taper, the immunosuppressive agent is continued at full dosage. The speed of the prednisone taper is determined by the level of disease activity. It is not necessary to have the disease totally suppressed before lowering the prednisone dose.
Patients who cannot take steroids may be treated with azathioprine, MMF, or cyclophosphamide alone. Patients who fail to respond to corticosteroids and immunosuppressive agents can be treated with IVIG, rituximab, plasmapheresis, or extracorporeal photopheresis.
Many patients with PF can be treated with potent topical steroids or low doses of corticosteroids. Hydroxychloroquine 200 mg twice a day was reported to be an effective adjuvant in patients with persistent and widespread PF. This was especially true when photosensitivity was present. The combination of nicotinamide (1.5 g/day) and tetracycline (2 g/day) or minocycline (100 mg twice a day) may be an effective alternative to steroids in PF and a steroid-sparing adjuvant, rather than a steroid alternative for PV. Dapsone may also be effective.
Sunlight exposure is harmful. Protection from sunlight should be part of the treatment.
Course and Remission
It is possible to eventually induce complete and durable remissions in most patients with pemphigus that permit systemic therapy to be safely discontinued without a flare in disease activity. The proportion of patients in whom this can be achieved increases steadily with time, and therapy can be discontinued in approximately 75% of patients after 10 years.
Risk of Relapse
There are three subtypes of PV: mucosal, mucocutaneous, and cutaneous. Compared to the other two types, the death rate is higher for the mucocutaneous type. Remission rates vary by subtype. Patients with the three different subtypes are treated with prednisolone 2 mg/kg/day plus azathioprine 2 to 2.5 mg/kg/day. The partial and complete remission rates, at the end of the first and second years of treatment, and the number of relapses have been compared in the three patient groups: 71.1% have mucocutaneous involvement, 18.8% have mucosal involvement, and 10.2% have only cutaneous involvement. The mean duration required for the mucocutaneous group to reach a prednisolone dosage of 30 mg/day is significantly longer. Mucocutaneous patients have a significantly lower rate of remission (31.9%) compared with those with only mucosal or cutaneous involvement (48.6%) at the end of the first year of the treatment. After 2 years, mucocutaneous patients have a lower remission rate (32.9% vs 44.5%). Relapses are more frequent in this subtype. Those presenting with mucosal or mucocutaneous erosions have a higher rate of active disease after receiving treatment for 1 year compared with those with only cutaneous presentation (66.7% vs 45%).
Conclusions
In the mucocutaneous subtype, clinical control is achieved later, and these patients have a lower rate of remission at the end of the first and second years of treatment. They are also prone to relapses.
The risk of disease relapse after treatment discontinuation in PV varies with the treatment regimen. Four therapy regimens have been compared: (1) oral prednisone at an initial dose of 100 mg (1.1 to 1.5 mg/kg/day) in monotherapy; (2) prednisone combined with oral azathioprine at a dose of 100 mg (1.1 to 1.5 mg/kg) per day; (3) prednisone combined with cyclosporin A at a dose of 2.5 to 3.0 mg/kg/day; or (4) prednisone combined with cyclophosphamide at a dose of 100 mg (1.1 to 1.5 mg/kg) per day. The risk of relapse is seen in 44% of patients. Treatment with prednisone plus oral cyclophosphamide combination therapy is associated with the lowest relapse rate and longest disease-free period; 54% of patients remain disease free for 5 years after treatment discontinuation.
Determining Remission and When to Stop Treatment.
Treatment is stopped when patients are clinically free of disease and when they have a negative finding on DIF. The titers of circulating antibodies have a rough correlation with disease activity, but they are not accurate enough to determine when to stop therapy. A skin biopsy for DIF can predict when a patient is in remission and may be used to predict relapse. A negative DIF finding suggests that there is immunologic remission, and 80% of patients with a negative DIF study remained disease free for the next 5 years.
Pemphigus in Association With Other Diseases
Myasthenia gravis and thymoma have been reported on many occasions in association with pemphigus (usually erythematosus and vulgaris). The clinical course is variable, but myasthenia gravis develops in most patients, followed by the detection of thymus disease, and finally by the appearance of pemphigus. Malignancy, usually of the lymphoid or reticuloendothelial systems, occurs more frequently in patients with pemphigus than in normal individuals. Paraneoplastic pemphigus is described in the following sections.
Drug-Induced versus Drug-Triggered Pemphigus
Drug-induced pemphigus presents with clinical and histopathologic features identical to the idiopathic form. Drugs implicated in pemphigus can be divided into two main groups according to their chemical structure: those containing a sulfhydryl radical (thiol drugs or sulfhydryl [SH] drugs) and nonthiol drugs. Penicillamine (a thiol drug) was the first drug reported to induce pemphigus ( Fig. 16.29 ![]() ). Pemphigus foliaceus has been reported in approximately 5% of patients taking 500 to 2000 mg of d -penicillamine or captopril for 2 months to 4 years. Most cases were mild. Patients with pemphigus induced by the SH drugs penicillamine and captopril showed spontaneous recovery in 39.4% and 52.6% of cases, respectively, once the drug was discontinued.
). Pemphigus foliaceus has been reported in approximately 5% of patients taking 500 to 2000 mg of d -penicillamine or captopril for 2 months to 4 years. Most cases were mild. Patients with pemphigus induced by the SH drugs penicillamine and captopril showed spontaneous recovery in 39.4% and 52.6% of cases, respectively, once the drug was discontinued.
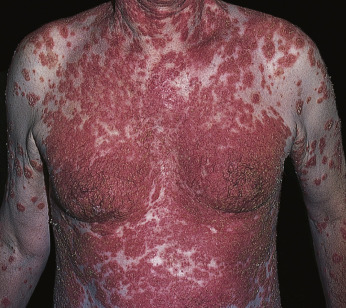
Pemphigus induced by other drugs shows spontaneous recovery in only 15% of cases. This suggests that penicillamine (SH drug) induces pemphigus, whereas other drugs only trigger the disease in patients with a predisposition.
The pemphigus-like eruption is not always limited, and the mortality approaches 10%.
The autoantibody response is similar in both spontaneous and drug-related disease. Therefore a similar molecular mechanism in the two types of pemphigus is suggested.
Paraneoplastic Pemphigus (Neoplasia-Associated Pemphigus)
Paraneoplastic pemphigus is an autoimmune disease that accompanies an overt or occult neoplasm and causes blisters. It has clinical and histologic features of both Stevens–Johnson syndrome and PV. There are painful mucosal ulcerations, conjunctival reactions, and polymorphous skin lesions on the trunk and extremities that usually progress to blisters ( Fig. 16.30 and Box 16.1 ). Antibodies against epithelial proteins are present in desmosomes and hemidesmosomes in the epidermis and respiratory epithelium. The prognosis is poor except for some patients who undergo total resection of their neoplasm. Progressive respiratory failure with clinical features of bronchiolitis obliterans is frequently the cause of death.
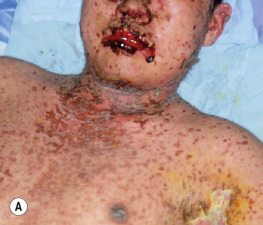
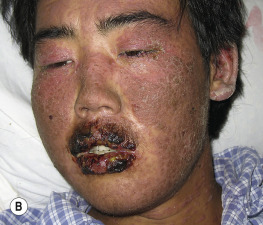
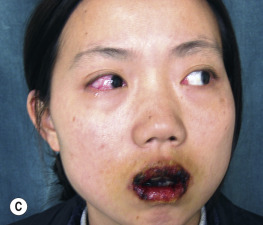
- •
Severe and painful mucosal involvement and polymorphic cutaneous eruptions
- •
Intraepidermal acantholysis, keratinocyte necrosis, and vacuolar interface dermatitis
- •
Indirect immunofluorescence of patient serum with rat bladder epithelia showing intercellular staining
- •
Presence of neoplasm, especially lymphoproliferative tumors
Laboratory Diagnosis of Paraneoplastic Pemphigus
Histologic Studies.
The histologic findings show features of pemphigus and erythema multiforme. There are intraepithelial clefts with epidermal acantholysis. In addition, dyskeratotic keratinocytes, vacuolar change of the basilar epidermis, and epidermal exocytosis of inflammatory cells also appear.
Direct Immunofluorescence.
Testing of mucous membrane and skin biopsies shows cell-surface deposits of IgG and C3 along with granular basement membrane deposits of C3.
When obtaining a specimen for testing, take a sample of normal mucosa that is ≈3 mm or more from a lesion or an area that demonstrates positive Nikolsky sign.
Indirect Immunofluorescence.
Indirect immunofluorescence with rat bladder substrate is used to differentiate PNP from classic pemphigus. Circulating IgG anti–cell surface and anticytoplasmic antibodies occur in a pattern and intensity unique to these patients.
The Pemphigoid Group of Diseases
Bullous pemphigoid, herpes gestationis, and cicatricial pemphigoid are autoimmune subepidermal blistering diseases with circulating IgG and BMZ–bound IgG antibodies and C3.
Bullous Pemphigoid
Bullous pemphigoid is the most common autoimmune blistering skin disease and is associated with an increased mortality. Like patients with other autoimmune diseases, patients with BP have an immune response to constituents of normal tissue. There is no racial or gender prevalence. Pemphigoid is a disease of the elderly, with most cases occurring after age 60, although cases have been reported in children. The mean age of onset is around 80 years. BP is associated with neurologic disease such as cerebrovascular disease, dementia, Parkinson disease, epilepsy, and multiple sclerosis. These conditions predate BP and are considered as risk factors. There is little evidence of an association of BP with internal malignancy. Drugs are often suspected of causing pemphigoid; stopping medication or changing to a different oral medication may help.
Clinical Manifestations.
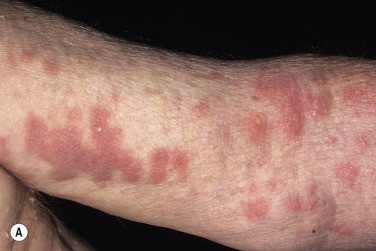
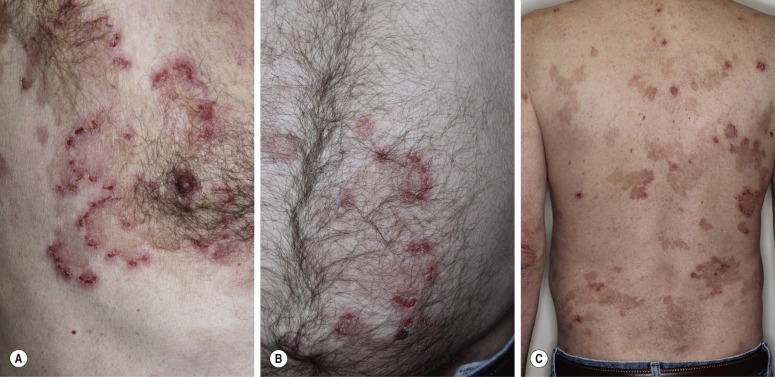
Oral blisters are present in 24% of patients. If present, they are mild and transient. Pemphigoid begins with a localized area of erythema or with pruritic urticarial plaques that gradually become more edematous and extensive. A diagnosis of hives is frequently made in this preblistering stage. The amount of itching varies but is usually moderate to severe. A group of elderly patients had pruritus for a mean period of 10 months before the diagnosis was made. In most cases the plaques turn dark red or cyanotic in 1 to 3 weeks ( Figs. 16.31 and 16.32 ), resembling erythema multiforme, as vesicles and bullae rapidly appear on their surface. Itching may be the only manifestation of the disease. Patients with itch without blisters have immunopathologic findings of BP.
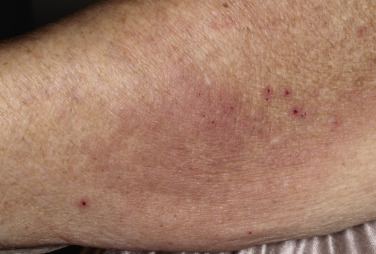
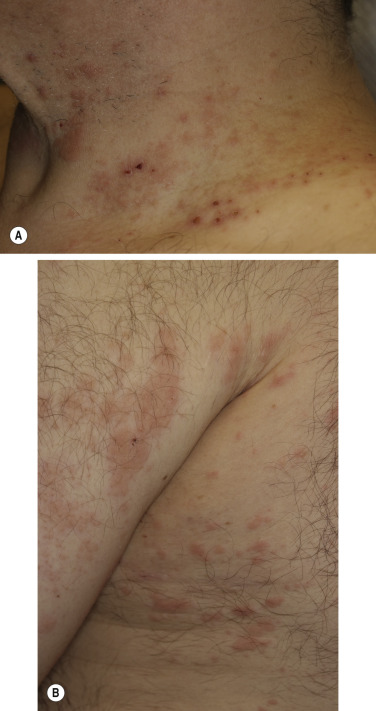
The eruption is usually generalized ( Figs. 16.33–16.35 ![]() ). The most common sites are the lower part of the abdomen, the groin, and the flexor surfaces of the arms and legs ( Fig. 16.36
). The most common sites are the lower part of the abdomen, the groin, and the flexor surfaces of the arms and legs ( Fig. 16.36 ![]() ). The palms and soles are affected. Involvement of genital mucous membranes occurs in 7% of patients. The 1- to 7-cm bullae appear isolated or in clusters and are tense with good structural integrity, in contrast to the large, flaccid, easily ruptured bullae of pemphigus. Firm pressure on the blister will not result in extension into normal skin as occurs in pemphigus; therefore Nikolsky sign is negative. Most bullae rupture within 1 week, leaving an eroded base that, unlike the situation with pemphigus, does not spread and heals rapidly.
). The palms and soles are affected. Involvement of genital mucous membranes occurs in 7% of patients. The 1- to 7-cm bullae appear isolated or in clusters and are tense with good structural integrity, in contrast to the large, flaccid, easily ruptured bullae of pemphigus. Firm pressure on the blister will not result in extension into normal skin as occurs in pemphigus; therefore Nikolsky sign is negative. Most bullae rupture within 1 week, leaving an eroded base that, unlike the situation with pemphigus, does not spread and heals rapidly.