Key Words
vascular tumors, cherry angioma, scleroderma, angiokeratoma, venous lake, pyogenic granuloma, Kaposi sarcoma, telangiectasia, infantile hemangioma, bacillary angiomatosis, lymphangioma circumscriptum, nevus flammeus, port-wine stain, Kasabach–Merritt syndrome
Congenital Vascular Lesions
A number of different congenital vascular lesions occur in the skin. The two major categories are vascular tumors (growths) and vascular malformations. Many represent developmental malformations and do not appear to be genetically determined, although the genetic basis for many vascular malformations is known. Vascular structures may be abnormal in size, abnormal in numbers, or both. These varied lesions have been referred to by many terms that have since been abandoned in favor of a simple classification, consisting of two groups, that is based on history and physical examination.
Hemangiomas of infancy (infantile hemangiomas [IH]) are the most common vascular growths of infancy and are vascular tumors composed of hyperplastic vascular endothelial cells that have the capacity for excessive proliferation but normally undergo eventual regression and involution. Vascular malformations are disorders of blood vessels due to localized defects of vascular morphogenesis and do not show markers of vascular proliferation. Vascular malformations result from structural abnormalities of mature endothelial cells that are usually present at birth and do not undergo hyperplasia with rapid proliferation. They have a normal pattern of growth and enlarge proportionally as the child grows. They do not resolve spontaneously. The distinction between a vascular malformation and a hemangioma of infancy may be difficult during the first few weeks of life. Infantile hemangiomas and vascular malformations have distinct natural histories ( Fig. 23.1 ).
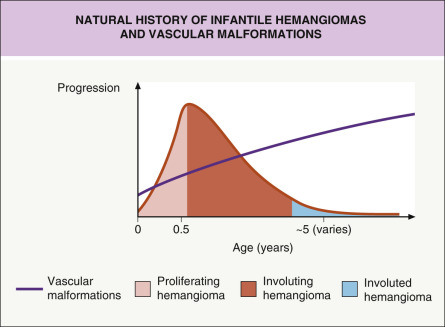
Hemangiomas of Infancy
Hemangiomas of infancy are benign neoplasms that result from rapid proliferation of endothelial cells. After an initial proliferating phase, many undergo complete regression with fibrosis. The color depends on its location. Hemangiomas of infancy may be classified as superficial, deep, mixed (superficial and deep), localized (focal), multifocal, or segmental (involving one or more embryologic segments). Hemangiomas involving the papillary dermis (superficial hemangiomas) are red; those in the reticular dermis and subcutaneous fat are blue or colorless (deep hemangiomas) ( Fig. 23.2 ). Large segmental hemangiomas may be associated with regional congenital anomalies – head and neck ( p osterior fossa malformations, h emangiomas (segmental), a rterial anomalies, c oarctation of the aorta, c ardiac defects, e ye abnormalities, s ternal defects, and s upraumbilical raphe [PHACES syndrome]) and lumbosacral ( l ower body segmental hemangioma, l ipoma, u rogenital anomalies, m yelopathy (tethered spinal cord), b ony anomalies, a norectal anomalies, a rterial anomalies, and r enal anomalies [LUMBAR syndrome]). All have the same vascular components and histopathology. Early hemangiomas are highly cellular, and numerous mast cells are present in the stroma. Lumina are more obvious and larger as the lesion matures. Vascular spaces may have features of capillaries, venules, and arterioles. Progressive interstitial fibrosis occurs during regression. There are subsets of congenital hemangiomas that grow rapidly (rapidly involuting congential hemangioma [RICH]), do not undergo involution (noninvoluting congenital hemangioma [NICH]), and partially involuting congenital hemangiomas.
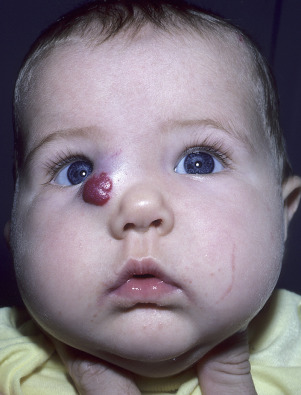
Superficial Hemangiomas of Infancy
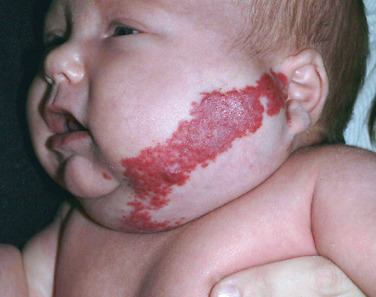
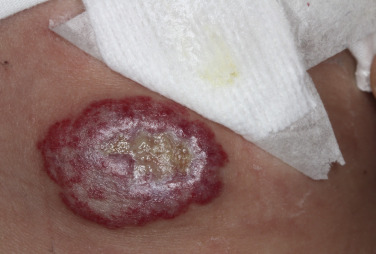
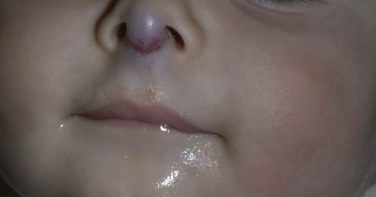
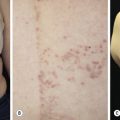
Superficial hemangiomas of infancy may be present at birth but more often appear within the first 2 weeks of life in 1% to 3% of infants; the female-to-male ratio is 3 to 1. Many children have one hemangioma, but 15% to 20% have several of these lesions. Most are small, harmless birthmarks that proliferate for 1 to 3 months and then slowly regress, over the next 3 to 5 years, leaving normal or slightly altered skin (see Fig. 23.1 ). They consist of a collection of dilated vessels in the dermis surrounded by masses of proliferating endothelial cells. These cells are responsible for the unique growth characteristics. The lesions begin as nodular masses or as flat, ill-defined, telangiectatic macules that are mistaken for bruises. Superficial hemangiomas grow rapidly from 5.5 to 7.5 weeks of age, forming nodular, protuberant, compressible masses of a few millimeters to several centimeters in diameter. This initial proliferative growth occurs in volume and not radial growth. Most IH reach maximal growth by 9 months of age. They are bright red with well-defined borders ( Fig. 23.3 ). Table 23.1 reviews risk features of hemangiomas of infancy and rationale for intervention. Vital structures can be compressed, and rapidly growing areas ( Fig. 23.4 ) may ulcerate. Larger lesions (>5 cm) ulcerate more frequently. Ulceration is the most common complication of IH ( Fig. 23.5 ![]() ). It can result in pain, infection, bleeding, and scarring and interfere with sleeping or feeding, all of which may adversely affect the quality of life of the child and the child’s family. Early white discoloration of IH is highly suggestive of impending ulceration.
). It can result in pain, infection, bleeding, and scarring and interfere with sleeping or feeding, all of which may adversely affect the quality of life of the child and the child’s family. Early white discoloration of IH is highly suggestive of impending ulceration.
| Risk Feature | Rationale for Intervention |
|---|---|
| HIGH RISK | |
| Segmental >5 cm on face | Associated structural anomalies (PHACES), scarring, visual/airway compromise, and gastrointestinal bleeding |
| Segmental >5 cm on lumbosacral/perineal area | Associated structural anomalies (LUMBAR), ulceration |
| Bulky lesion on face (prominent dermal thickening, steep ascent from normal to involved skin) | Tissue distortion with risk of permanent scarring/disfigurement |
| Early white discoloration | Marker of ulceration |
| Central face | High risk for disfigurement |
| Periorbital, perinasal, and perioral | Functional compromise (visual, respiratory, and nutrition), risk of disfigurement |
| Multiple hemangiomas >5 | Gastrointestinal bleeding and visceral hemangiomas |
| INTERMEDIATE RISK | |
| Lateral face, scalp, hands, and feet | Risk of disfigurement lower, but possible risk of functional compromise |
| Body folds (neck, perineum, and axillae) | Higher risk of ulceration |
| Segmental >5 cm – trunk, arms, and legs | Risk of ulceration and permanent residual skin changes |
| LOW RISK | |
| Trunk, arms, and legs (nonvisible) | Low risk of disfigurement or functional compromise |
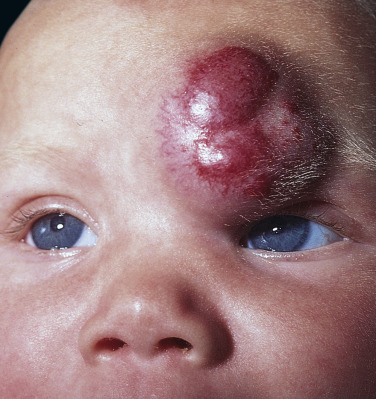
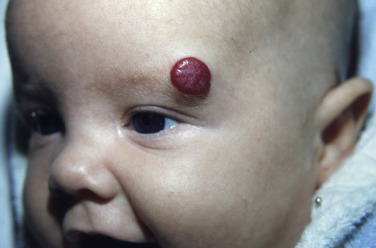
Hemangiomas may block vision, interfere with feeding or respiration, or obstruct the external auditory canal. Recurrent bleeding may complicate ulceration. Most have a benign course ( Figs. 23.6 and 23.7 ). An inactive phase lasting several months is followed by fibrosis and involution. The involution phase ends around 3 years of age ( Figs. 23.8 and 23.9 ).
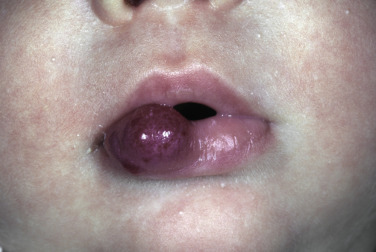
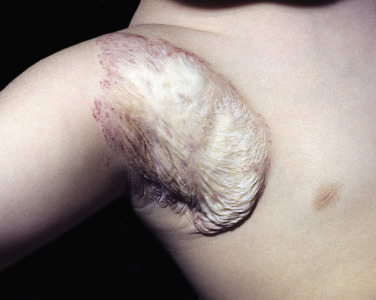
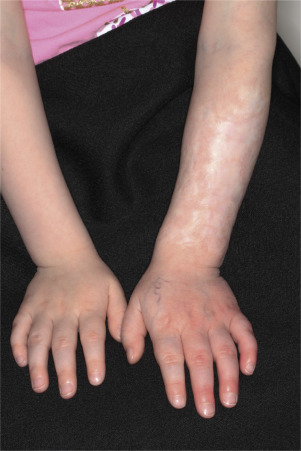
Deep Hemangiomas
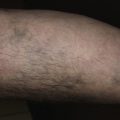
Deep (cavernous) hemangiomas are collections of dilated vessels deep in the dermis and subcutaneous tissue that are present at birth. Localized and superficial venous lesions may coexist with venous ectasias and deep vein anomalies. Clinically they appear as pale, skin-colored, red, or blue masses that are ill-defined and rounded ( Figs. 23.10 to 23.14 ![]() ). Most are asymptomatic. Hyperhidrosis over the lesion is common, and often there are recurrent episodes of thrombophlebitis in or near the lesions. Like superficial hemangiomas, the lesions enlarge for several months, become stationary for an indefinite period, and undergo spontaneous resolution. The growth phase of deep, mixed, and segmental hemangiomas of infancy may be delayed by a month and proliferate a month longer compared to superficial hemangiomas of infancy. They are managed in the same manner as superficial hemangiomas.
). Most are asymptomatic. Hyperhidrosis over the lesion is common, and often there are recurrent episodes of thrombophlebitis in or near the lesions. Like superficial hemangiomas, the lesions enlarge for several months, become stationary for an indefinite period, and undergo spontaneous resolution. The growth phase of deep, mixed, and segmental hemangiomas of infancy may be delayed by a month and proliferate a month longer compared to superficial hemangiomas of infancy. They are managed in the same manner as superficial hemangiomas.
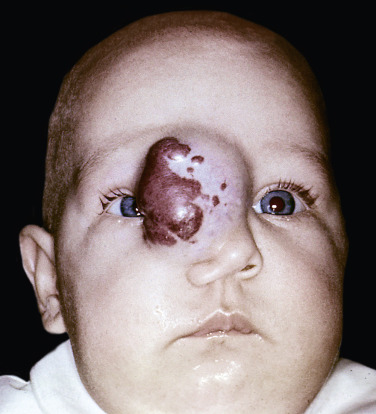
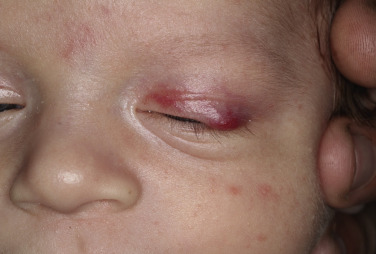
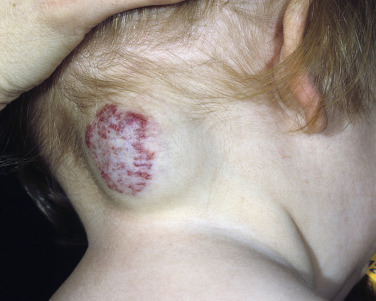
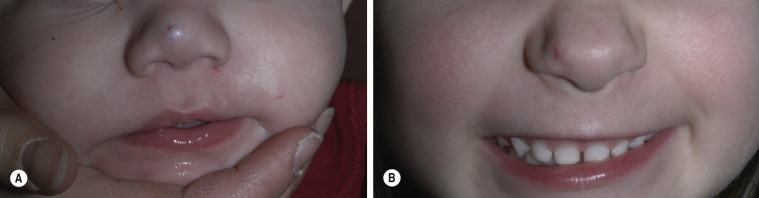
Location, Number, and Risk.
Hemangiomas of infancy in certain locations may be associated with other anomalies or have a risk of developing complications (see Table 23.1 ). Most complications arise during the proliferative growth phase. Infants with greater than five hemangiomas are at risk for visceral hemangiomas, especially involving the liver. Hemangiomas involving the facial “beard” area are associated with subglottic hemangiomas. Segmental hemangiomas may be associated with structural anomalies and hypothyroidism (see Table 23.3 ).
Management.
Most parents and families are very concerned when their child has a hemangioma of infancy. Providers must be well versed in the natural history, complications, and management of these common vascular growths. The vast majority of hemangiomas of infancy are uncomplicated and will resolve without adverse sequela. Close monitoring with photography and follow-up visits are very helpful. Newborns and infants less than 3 months of age should be seen weekly to every other week since rapid growth occurs earlier in life. Older infants, in whom the hemangioma growth has slowed, can be monitored monthly or less often . During these visits, the size of the hemangioma should be monitored and patients should be evaluated for signs of ulceration and cosmetic or functional complications.
Infants with high-risk hemangiomas (see Table 23.1 ) should undergo appropriate imaging and specialty referrals. See flow diagram for complicated and uncomplicated hemangiomas ( Fig. 23.15 ![]() ).
).

Ulcerated Hemangiomas.
Ulcerated hemangiomas are very painful and must be treated early. Table 23.2 summarizes treatment for ulcerated hemangiomas. Keeping the wound covered with a dressing or thick emollient is essential to minimize the pain associated with ulcerated hemangiomas.
| Wound Care | Adjuvant Therapies |
|---|---|
|
|
Medical Therapy
Beta-Blockers.
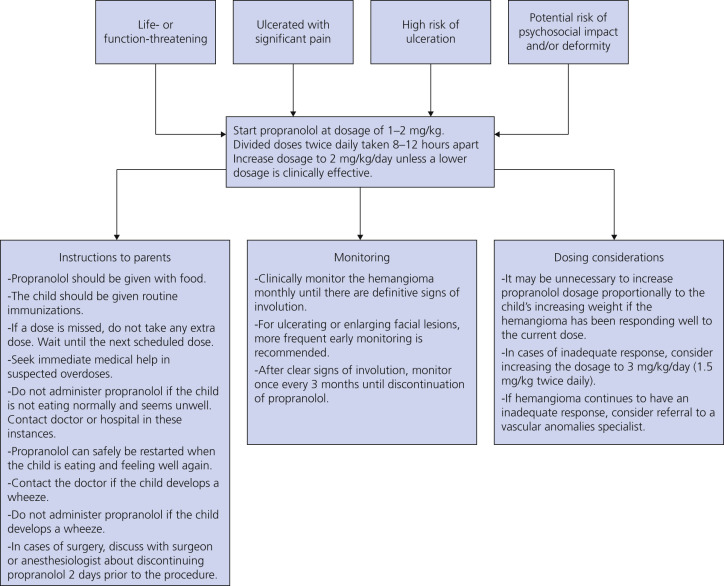
Propranolol is a nonselective beta-blocker and is considered to be the first-line agent for hemangiomas of infancy. Table 23.3 lists indications and contraindications for propranolol use in hemangiomas of infancy. Propranolol may be applied topically (propranolol 1%, must be compounded in the United States) or taken systemically. Most infants do not require extensive pretreatment evaluation, other than a complete physical examination. If there is a history of cardiac and pulmonary disease, then appropriate consultations should be completed before starting propranolol. Infants with concern for central nervous system vascular shunting, such as those with PHACES syndrome, should have head and neck imaging with magnetic resonance imaging (MRI)/magnetic resonance angiogram (MRA) before starting propranolol. Propranolol is started in the inpatient setting for premature and high-risk infants. Older term infants may be started on propranolol in the outpatient setting, but require glucose and blood pressure monitoring. Propranolol has been shown to result in complete or near complete remission in 60% of patients when treated for 6 months with minimal side effects. Patients should be followed at 1- to 3-month intervals (sooner for newborns and infants less than 1 month of age). Patients are treated for 6 to 12 months and in some instances longer. Rarely infants may experience rebound growth. Instructions to parents and monitoring and dosing considerations are summarized in Fig. 23.16 .
| Indications | Contraindications |
|---|---|
|
|
Topical Beta-Adrenergic Blockers.
Small superficial hemangiomas may be treated with topical beta-blockers (propranolol 1%, timolol 0.5%). Topical beta-blockers are applied 2 times daily. Topical therapy may result in improvement in size, color, and volume in over 70% of patients after 6 to 9 months of treatment. Rarely, bradycardia may occur in small preterm infants. Topical beta-blockers are an excellent choice for small hemangiomas or those in cosmetically sensitive sites such as the face.
Corticosteroids.
Topical corticosteroids (such as clobetasol, mometasone, and triamcinolone) may be utilized to treat small superficial hemangiomas, but are now used less frequently because of the effectiveness of topical beta-blockers. Intralesional steroids (triamcinolone acetonide 10 to 40 mg/cc, not to exceed 3 mg/kg) injected monthly may be utilized for deep bulky localized hemangiomas. Systemic corticosteroids are utilized in patients with complicated hemangiomas who are unable to take propranolol. Prednisolone is dosed at 2 to 3 mg/kg/day as a single morning dose. Patients are treated for 4 to 12 weeks at the full dose and then tapered to end treatment by 9 to 12 months of age. Tapering too soon results in rebound growth of the hemangioma. Table 23.4 ![]() summarizes the indications, formulations, treatment schedules, and side effects of topical, intralesional and systemic corticosteroids. Guidelines for the use of intralesional steroids in periorbital hemangiomas are shown in Box 23.1
summarizes the indications, formulations, treatment schedules, and side effects of topical, intralesional and systemic corticosteroids. Guidelines for the use of intralesional steroids in periorbital hemangiomas are shown in Box 23.1 ![]() .
.
| Treatment Method | Formulations | Treatment Schedules | Potential Side Effects |
|---|---|---|---|
| Systemic | Prednisolone |
|
|
| Intralesional |
|
|
|
| Topical |
|
|
|
Evaluation
History and physical examination
Computed tomography scan
Avoidance of immunization with live virus vaccines
Short-acting, light, general anesthetics
Pharmacologic Agent
Triamcinolone 10 to 20 mg/mL, with a maximum injection of 3 to 5 mg/kg per procedure
Procedure
Anterior approach to the eyelid is preferred
Multiple injection sites: 0.1 mL aliquot
Aspirate before injecting (27- or 30-gauge needle)
Digital pressure is then applied to avoid hematomas
Repeat up to three times at 8-week intervals or until regression has ceased
Other medical treatments such as vincristine, interferon-α, and imiquimod are utilized in specialized or recalcitrant cases. In the future, newer anti-antiangiogenic agents may be utilized to treat hemangiomas of infancy.
Other Treatments.
Laser (pulsed dye laser) has been utilized extensively in the past to treat hemangiomas of infancy. At the present time, pulsed dye laser is utilized as part of a multimodal treatment regimen, specifically for early superficial hemangiomas, superficial and deep hemangiomas (special locations such as the nasal tip) to help preserve the overlying skin, refractory ulceration, and residual telangiectasia or partially involuted hemangioma with telangiectasia. Hypertrophic scarring and hypopigmentation are potential complications of laser treatment.
Surgery for hemangiomas of infancy may be considered in select patients. Patients are considered candidates for surgery if they have failed or are unable to undergo medical treatment, the hemangioma of infancy is easily removed with limited cosmetic deformity, and if the timing of surgery would not impact the scar. Elective surgery may be performed after age 4, before psychosocial influences take hold and after the hemangioma involutional phase. Table 23.5 ![]() highlights indications and clinical considerations for special anatomical sites.
highlights indications and clinical considerations for special anatomical sites.
| Indications |
|
| Timing of surgical intervention |
|
| Anatomical considerations |
|
Kasabach–Merritt Phenomenon (KMP).
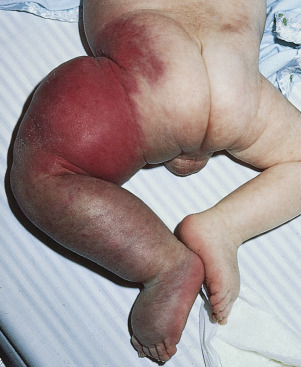
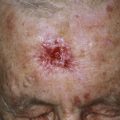
Kasabach–Merritt phenomenon is a variant of disseminated intravascular coagulation (DIC) in which platelets and clotting factors are locally consumed within two rare tumors: kaposiform hemangioendothelioma (70%) and tufted angioma (10%) ( Table 23.6 ![]() ). Although described as two separate entities, these rare tumors are very similar in clinical appearance and may be part of a continuum with superficial lesions showing findings of tufted angioma and deeper lesions infiltrating into the subcutis and viscera showing features of kaposiform endothelioma. Both vascular lesions arise from capillary and lymphatic endothelium and share an identical immunophenotype (both stain positive for Prox-1, D2-40, LYVE1, CD31, and CD34). KMP should be suspected in any infant with rapid painful enlargement of a vascular tumor. The vascular tumor will become tense, bruised, and painful ( Fig. 23.17 ). There is a simultaneous decrease in the platelet count (3000–60,000/microL), fibrinogen less than 1 g/L, and elevated D-dimer and fibrin degradation products. Prothrombin and activated partial thromboplastin times are normal or slightly elevated. Patients may develop severe anemia from sequestration of red blood cells in the tumor. The blood smear may show microangiopathic hemolytic anemia. These hematologic alterations are thought to occur because of intralesional platelet trapping and fibrinogen consumption. Generalized petechiae occur when platelet counts drop below 10,000 per microliter. Despite these hematologic anomalies, bleeding is rare. Box 23.2 lists the initial evaluation of an infant with suspected KMP. Table 23.7 summarizes the hematologic, surgical, and pharmacologic support of KMP. The mortality rate of KMP is as high as 24%, but has likely improved with the combination of hematologic, surgical, and pharmacologic treatment.
). Although described as two separate entities, these rare tumors are very similar in clinical appearance and may be part of a continuum with superficial lesions showing findings of tufted angioma and deeper lesions infiltrating into the subcutis and viscera showing features of kaposiform endothelioma. Both vascular lesions arise from capillary and lymphatic endothelium and share an identical immunophenotype (both stain positive for Prox-1, D2-40, LYVE1, CD31, and CD34). KMP should be suspected in any infant with rapid painful enlargement of a vascular tumor. The vascular tumor will become tense, bruised, and painful ( Fig. 23.17 ). There is a simultaneous decrease in the platelet count (3000–60,000/microL), fibrinogen less than 1 g/L, and elevated D-dimer and fibrin degradation products. Prothrombin and activated partial thromboplastin times are normal or slightly elevated. Patients may develop severe anemia from sequestration of red blood cells in the tumor. The blood smear may show microangiopathic hemolytic anemia. These hematologic alterations are thought to occur because of intralesional platelet trapping and fibrinogen consumption. Generalized petechiae occur when platelet counts drop below 10,000 per microliter. Despite these hematologic anomalies, bleeding is rare. Box 23.2 lists the initial evaluation of an infant with suspected KMP. Table 23.7 summarizes the hematologic, surgical, and pharmacologic support of KMP. The mortality rate of KMP is as high as 24%, but has likely improved with the combination of hematologic, surgical, and pharmacologic treatment.
Laboratory studies
CBC w/platelet count
Coagulation studies (PT, PTT, fibrinogen, d-Dimer levels)
Imaging studies
MRI with and without contrast
Tissue biopsy
CBC, complete blood count; MRI, magnetic resonance imaging; PT, prothrombin time; PTT, partial prothrombin time.
| Hematologic | Surgical/Radiologic | Pharmacologic | Duration of Pharmacologic Therapies | Supportive |
|---|---|---|---|---|
|
|
|
|
|
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


