Vascular Malformations: Introduction
|
Epidemiology
Vascular malformations are believed to be due to errors of development of vessels that occur during the fourth and tenth weeks of intrauterine life. Most vascular malformations are sporadic, although several families with inherited forms have been identified. They are very heterogeneous and affect about 0.3% of the population. Vascular malformations are mostly congenital, even if they may be diagnosed only later in life.
Etiology and Pathogenesis
Although several genes have been identified as the cause of inherited forms of vascular malformations, the etiology of the most common sporadic lesions is unknown.1–3 Histologically, vascular malformations consist of enlarged, tortuous vessels with quiescent endothelium. In contrast to hemangioma, there is neither parenchymal mass nor cellular proliferation.
Classification
For many years, vascular anomalies were grouped under the term angioma, hampering precise classification and leading to incorrect diagnosis and improper management. For example, the term hemangioma has been used both for vascular malformations, often of venous type (cavernous hemangioma) as well as for vascular tumors (strawberry hemangioma). This nomenclature changed in 1982 with the development of a biologic classification by Mulliken and Glowacki. It divided vascular anomalies into two major categories: (1) vascular tumors (with cellular proliferation; hemangioma being the most common) (see Chapter 126), and (2) vascular malformations (structural anomalies of blood vessels) that are subsequently subdivided, depending on the affected vessel type, into arterial, capillary, lymphatic, or venous malformations.4,5 In 1996, this classification was adopted and further developed by the International Society for the Study of Vascular Anomalies (ISSVA) (Table 172-1).6 Vascular malformations mostly affect only one vessel type, yet combined malformations also exist. They are named according to the affected vessel types, (e.g., capillary-venous or venolymphatic malformation). In addition to isolated forms, vascular malformations occur in syndromes such as Klippel–Trenaunay (KT) (capillary-lymphaticovenous malformation with limb hypertrophy), Maffucci syndrome (multiple enchondromas associated with multiple venous anomalies and high incidence of malignancy)7, Cloves syndrome (congenital lipomatous overgrowth with vascular malformations, epidermal nevi, and scoliosis)8,9, or Parkes Weber syndrome (high-flow vascular malformation of the extremity with soft tissue hypertrophy).10
Vascular Tumors | Vascular Malformations |
|---|---|
Hemangioma | Capillary |
Hemangioma of infancy (HOI) | Capillary malformation (CM) (Port-wine stain) |
Congenital | Telangiectasia (hereditary benign telangiectasia, essential telangiectasia) |
Rapidly involuting congenital hemangioma (RICH) | Hereditary hemorrhagic telangiectasia (HHT) |
Noninvoluting congenital hemangioma (NICH) | Capillary malformation-arteriovenous malformation (CM-AVM) |
Sturge–Weber syndrome | |
Hemangioendotheliomas | Venous |
Kaposiform hemangioendothelioma | Venous malformation (VM) |
Tufted angioma | Familial form: cutaneomucosal Mucocutaneous VM venous malformation cutaneomucosal (VMCM) |
Glomuvenous malformation (GVM) | |
Blue rubber bleb nevus or Bean syndrome (BRBN) | |
Angiosarcoma | Lymphatic |
Lymphatic malformation (LM) | |
Primary Lymphedemas | |
Arterial | |
Arteriovenous malformation (AVM) | |
Capillary malformation-arteriovenous malformation (CM-AVM) | |
Arteriovenous fistula (AVF) | |
Syndromic Malformations | |
Slow-flow | |
Klippel–Trenaunay syndrome CLVM1 (capillary-lymphaticovenous malformation with limb hypertrophy) | |
Maffucci syndrome CLOVES syndrome | |
Fast-flow | |
Parkes Weber syndrome |
Clinical Findings
Vascular malformations grow proportionately with the patient. Usually they do not regress. Most frequently, they are well demarcated and localized (Fig. 172-1). In rare instances, they can be the stigmata of deep lesions. Vascular malformations are rheologically divided into slow-flow (capillary, lymphatic, venous, and combined) and fast-flow (arterial, arteriovenous, and combined). Doppler ultrasound is the best noninvasive radiologic examination that provides clues into the differentiation of the various types. Magnetic resonance imaging (MRI) details the extension and precise location of the lesion (Table 172-2).11
Figure 172-1
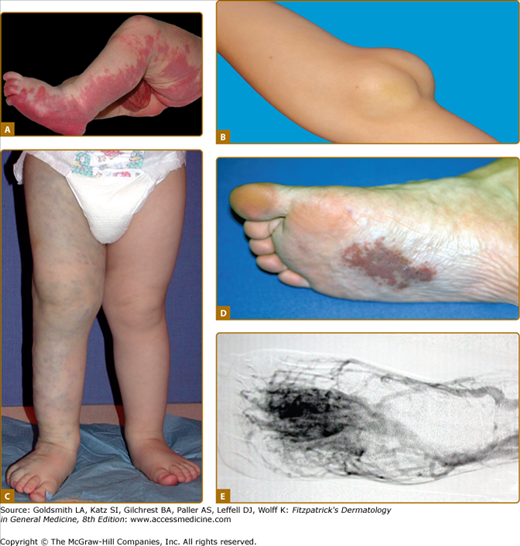
Vascular malformations of various vessel types: A. capillary malformation of left lower extremity and genitalia; B. subcutaneous lymphatic malformation invading the cubital nerve; C. extensive venous malformation of right lower extremity causing pain, swelling, and coagulopathy; D. arteriovenous malformation of sole; and E. note nidus of the lesion on arteriography.
Capillary Malformation | Venous Malformation | Lymphatic Malformation | Arteriovenous Malformation | |
|---|---|---|---|---|
Skin color | Pinkish-red to purple | Normal-bluish-purple GVM: red (neonates) to dark-blue-purple (adult) | Normal-yellowish-purple | Normal-red |
Aspect | Flat | Flat-raised | Raised-vesicular | Flat-raised |
Temperature | Normal | Normal | Normal | Warm |
Palpation | Normal | Compressible GVM: nodular, painful | Firm, non compressible | Thrill, bruit |
Associations | Hypertrophy pyogenic granulation | Phleboliths, deformation | – | Ulceration, deformation |
Radiology | No-flow | Slow-flow | Slow-flow, cyst | Fast-flow |
Histology | Dilated capillaries D2–40 negative | Thin-walled venous channels Relative lack of smooth muscle cells D2–40 negative | Cystic lymphatic channels D2–40 positive | Arterialized veins D2–40 negative |
Etiology | ? | VMCM: TIE2 GVM: Glomulin Sporadic VM: 50% somatic TIE2 mutations | LM: ? Primary lymphedema: SOX18, VEGFR3, FOXC2, CBR2 | AVM: ? CM-AVM: RASA1 HHT: endoglin, Activin-like receptor tyrosine kinase, SMAD4 PTHS: PTEN |
Treatment | Pulsed-dye laser | Sclerotherapy, surgery | Surgery, sclerotherapy | Embolization followed by surgical resection of nidus |
Treatment
Treatment of vascular malformations depends on the affected vessel type, the location of the lesion, and the symptoms. As many lesions are extensive, patients should be aware that a complete cure is often not possible. Treatment is difficult and can have severe complications. Extensive and/or complex lesions should always be managed by a multidisciplinary team.
Capillary Malformations (CM)
|
Capillary malformation (CM), commonly called port-wine stain, is a slow-flow vascular malformation with an incidence of 0.3% (Fig. 172-2).12 Other blanchable pinkish patches, known as stork bite, angel’s kiss, salmon patch, or nevus flammeus neonatorum, are often confused with CM. They are located on the nape of the neck (81%), the eyelids (45%), or the glabella (33%).13 When located on the occiput, it is called Unna nevus (see eFig. 172-2.1). These patches have a much higher incidence (42%) in white infants than in black infants (31%).14 They are also present in various syndromes such as Beckwith–Wiedemann and Rubinstein–Taybi. These lesions disappear spontaneously around the ages of 1–4 years.
Figure 172-2
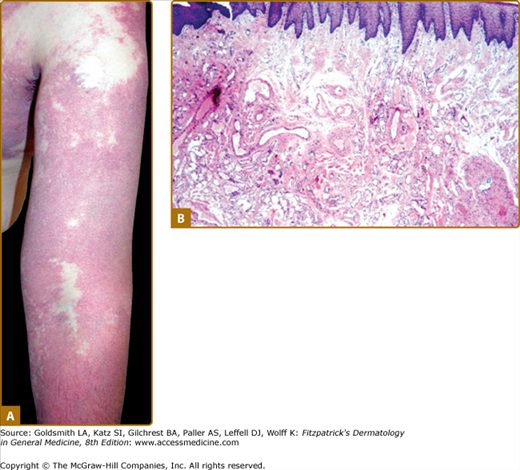
A. Extensive capillary malformation of upper extremity. B. Histologically, capillary malformation is characterized by dilated capillaries of normal number on the papillary and upper reticular dermis in combination with areas of increased number of normal-looking capillaries (hematoxylin and eosin staining).
CM are mainly sporadic, although there are well-documented pedigrees showing autosomal dominant inheritance.15 When inherited, they are usually multiple and part of the CM-AVM phenotype, that associates atypical CM with arteriovenous malformation (AVM) (see Section “Arteriovenous Malformations”).16–19 No sex preponderance is noted.
Etiology of CM is currently unknown. Facial CM is thought to be due to clonal expansion of abnormal cells originating from the neural crest.20 The identification of the affected gene (RASA1) in CM-AVM (see Section “Arteriovenous Malformations”) might help unravel the cause of the more common sporadic CM. Histologically, CM is characterized by dilatation of normal numbers of capillaries of the papillary and upper reticular dermis combined with areas of increased number of normal-looking capillaries (see Fig. 172-2). Endothelial cells are flat. Factor VIII, fibronectin, and basement membrane protein are normal but S100 staining shows abnormal innervation.21,22
(see Figs. 172-2 and 172-3). CM is a red homogenous congenital lesion that is often unilateral, sometimes bilateral, but usually not median. CMs involve skin and subcutis, and sometimes mucosa. Their color varies from pinkish red to deep purple with a geographic contour or a dermatomal distribution. Lesions are flat and painless, do not bleed spontaneously, and are never warm on palpation. They can extend throughout the entire body. Fifty percent of CMs are located on the face where they follow the distribution of the trigeminal nerve: ophthalmic branch V1 (front and upper eyelid), maxillary branch V2 (lower eyelid, cheek, and upper lip) (see Fig. 172-3B), or mandibular V3 (lower lip, chin, and mandibule). In combination with another vascular malformation, CM can be part of a syndrome, such as Sturge–Weber, phakomatosis pigmentovascularis (PPV), Klippel–Trenaunay (KT), Servelle–Martorell, etc. None of these syndromes is inherited. In rare instance, CM can be the cutaneous hallmark of occult spinal dysraphism, especially if located in the lumbosacral area.23
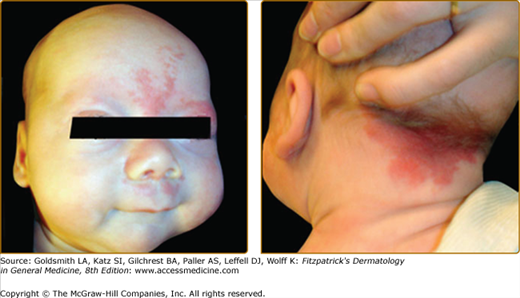
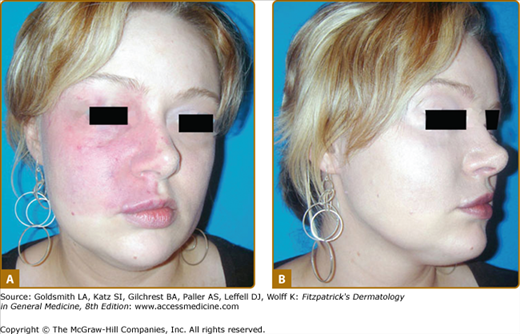
Sturge–Weber Syndrome (SWS) is a neuro-oculocutaneous syndrome consisting of a CM located on the opthalmic branch of the trigeminal nerve, a homolateral leptomeningeal capillary-venous malformation, often on the parieto-occipital area, and choroid “angioma” causing glaucoma and sometimes buphthalmos and retinal detachment (Fig. 172-4). Calcifications are frequently seen on the choroid plexus, and anomalies of the venous drainage of the encephalon can occur.24 This malformation can cause epilepsy, leading to neural death, cerebral atrophy, and mental retardation.
Figure 172-4
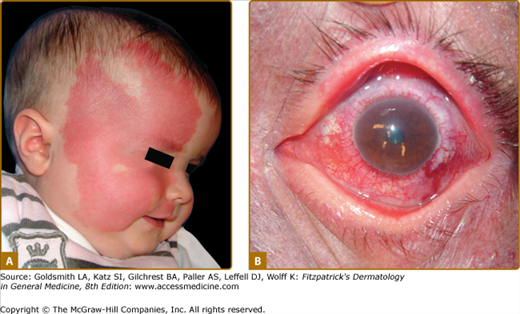
Extensive right facial capillary malformation (A) involving the ophthalmic and maxillary branches of the trigeminal nerve in a 6-month-old girl affected with Sturge–Weber syndrome. Choroid capillary-venous malformation (B) causing vision loss in a 50-year-old man with Sturge–Weber syndrome.
Phakomatosis pigmentovascularis (PPV) is thought to be an embryogenic anomaly affecting the vasomotor nerves and the melanocytes, both derived from neural crest. Clinically, PPV manifests as a large capillary malformation, which is metameric in distribution and mainly located on the trunk or the extremities, in association with pigmented cutaneous lesions, such as a pigmented nevus, a nevus spilus, a café au lait patch, or an atypical Mongolian spot (see Chapter 75) that is not located on the sacrum (eFig. 172-4.1).25 Nevus anemicus can also be seen in the vicinity as a twin spot.26 These cutaneous lesions can be associated with systemic, visceral (e.g., larynx hypoplasia, intestinal polyposis), muscular (scoliosis), neurologic (mental retardation, epilepsy, intracranial calcification, cerebral atrophy), or ocular involvement (anomaly of the iris).
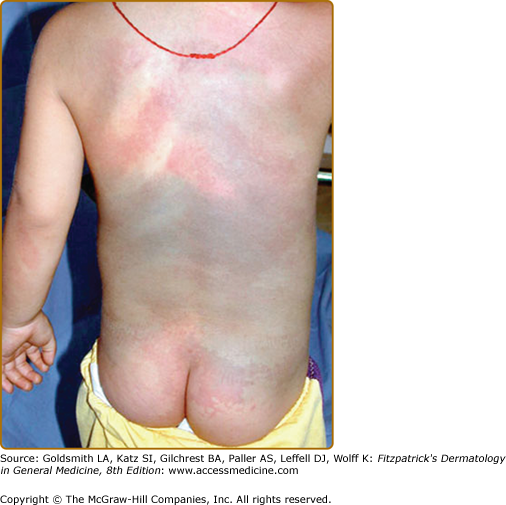
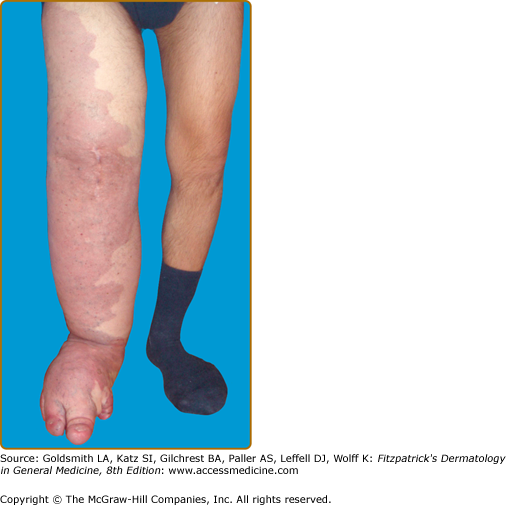
Klippel–Trenaunay syndrome (KTS) is an eponym used for the presence of a capillary-lymphaticovenous malformation located on the extremity with hypertrophy of the affected limb (Fig. 172-5). Venous varicosities on the lateral side of the leg, first described by Servelle, are very common and is specific for this syndrome in 80% of cases.27 It is thought to be due to the persistence of embryologic veins. Valvular incompetence of deep venous system is common. Lower extremities are most commonly affected (95%), although upper limb, thorax, abdomen, and genitalia can also be involved. Lymphatic dermal vesicles cause frequent oozing. Lymphedema can be seen and tends to progress with time. Infection is common after trauma or secondary to small ulceration. VM is often seen in the underlying muscles. Hypertrophy of the limb is another sign of this disorder. It is already present at birth and occasionally evolves after puberty. Evolution toward ulceration, infection, bleeding, and thrombosis is common. Protein-loosing enteropathy can be seen with abdominal involvement. Pulmonary embolism can sometimes lead to death.
No imaging studies are mandatory except in some rare situations. If a so-called CM is painful, warm, or spontaneously bleeds, Doppler ultrasound is indicated to exclude the diagnosis of a fast-flow malformation, such as an AVM, a Parkes Weber syndrome, or a proliferating hemangioma. CM located in the frontopalpebral area, especially if the inner part of the upper eyelid is involved, can be part of SWS. Therefore, a brain MRI and an ophthalmic examination have to be performed during the first months of life and repeated once a year until puberty, even if normal at younger age.28 MRI of the spinal cord is needed in presence of a lumbosacral CM. In patients with PPV, ophthalmologic, neurologic, and orthopedic follow-up is mandatory due to the common association of CM with systemic lesion. In patients with KTS, leg length is evaluated radiologically after the age of 2 years and, if abnormal, followed once a year until the end of puberty. Doppler ultrasound evaluation of the venous status is mandatory to identify varicose veins in KTS. MRI is sometimes useful before management of the lesion.27
Differential Diagnosis of CM | Differential Diagnosis of CM (site specific) |
|---|---|
Most Likely Faint pinkish median patch
Multiple, inherited
Telangiectatic patch Warm on palpation
Ulcerated or painful
Rule Out Small, multiple
| Most Likely Occiput
Glabella, upper eyelid, nape of neck
Consider Extremity
Faint on trunk
Rule Out Scalp |
| aCMTC = Cutis Marmorata Telangiectatica Congenita; HHT = Hereditary Hemorrhagic Telangiectasia; M-CM = Macrocephaly-Capillary Malformation | |
AVF = arteriovenous fistula; AVM = arteriovenous malformation; CM = capillary malformation; CM-AVM = capillary malformation-arteriovenous malformation; CMTC = cutis marmorata telangiectatica congenita; HHT = hereditary hemorrhagic telangiectasia; M-CM = macrocephaly cutis marmorata
The major concern for a patient affected with CM is cosmetic due to the visible discoloration. Hypertrophy of soft tissue can occur with time, especially when CM is located on V2 and V3 dermatomes or on the extremities. In rare instances, CM can be the stigmata of underlying anomalies such as lumbar dysraphism when located at sacrum.
CM never regresses. It is present at birth, and slowly thickens and darkens with time. It often becomes raised and nodular.35 Pyogenic granuloma can occur with time as well as soft tissue or bony hypertrophy.36 (Fig. 172-6).
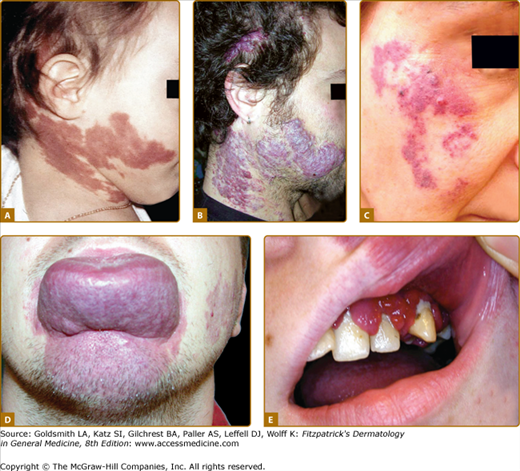
Many therapeutical approaches have been used for CM, such as electrocoagulation, tattooing, dermabrasion, cryosurgery, and cosmetic makeup. Camouflage using Covermark is still effectively used (see eFig. 172-6.1). Laser treatment is the gold standard for most CM. Pulsed-dye laser with its specific wavelength (585 nm) and a short pulse duration (400 ms) currently gives the best results in infants and children. There are few complications. Multiple sessions (6–12) are needed, and general anesthesia may be necessary as the procedure is painful. Laser treatment is more efficient on the cervicofacial and trunk area than on the extremities. Early treatment during childhood does not reduce the number of laser sessions.37 The use of a dynamic cooling system to avoid heating the epidermis allows increasing of laser fluences, resulting in optimal lightening of the lesion.38 Recurrence can occur after cessation of therapy. Laser has no impact on associated hypertrophy. Contour resection is needed mainly to treat complications such as pyogenic granuloma or lip hypertrophy.
Treatment of patients with KTS is conservative and mainly consists of elastic stockings to alleviate pain due to venous congestion. Surgical resection of varicose veins cannot be done if the deep venous system is abnormal (aplasia, incompetence). In the presence of leg discrepancy, an early-adapted shoe lift is needed to prevent scoliosis, and is followed by epiphysiodesis. If cutaneous bleeding occurs from capillary-lymphatic cutaneous vesicles, neodymium:yttrium-aluminum-garnet laser coagulation, sclerotherapy, or skin grafting can be helpful.
In patients with SWS, ophthalmic follow-up, started immediately after birth and continued until the end of puberty, is essential, as visual impairment will depend on the promptness of treatment. Prophylactic antiepileptic medication to prevent neural cell death has been advocated by some clinicians.39,40
Venous Anomalies
|
Venous anomalies are the most common vascular malformations referred to specialized center 41. They are mainly sporadic, although 1% are inherited (VMCM) and 5% are inherited glomuvenous malformations (GVM). No sex preponderance is reported.42 Both VMCM and GVM have an age-dependant variation in penetrance, which reaches its maximum by 20 years of age (87% for VMCM and 92.7% for GVM).43 Large VMCMs and GVMs are present at birth. However, 17% of affected individuals develop new small lesions over time.42
VM is a congenital defect on the skin or mucosa, but can involve any structure (subcutis, muscles, bones, and nerves) and any organ [central nervous system, gastrointestinal (GI) tract]. Fifty percent of VMs are located in the cervicofacial area and 37% on the extremities. VMs are mainly isolated, but can be part of complex vascular disorders of unknown etiology, such as KTS, and Maffucci and blue rubber bleb nevus (BRBN) syndromes.44
VMs consist of ectatic vascular channels of venous type with flat endothelium and thin walls due to a variable number of mural smooth muscle cells that stain positive for smooth muscle cell-α-actin.45 In contrast, GVM is characterized by distended venous channels surrounded by mural glomus cells that are aberrant smooth muscle cells. Glomus cells are round or polygonal, instead of elongated like the normal vascular smooth muscle cells.46 By immunohistochemistry, glomus cells stain positively for smooth muscle-α-actin and vimentin, whereas they are negative for desmin, von Willebrand factor and S100 neuronal marker.47 By in situ hybridization, they are also negative for glomulin.48
The etiopathogenesis of at least 50% of sporadic VM are due to somatic mutations in the tyrosine kinase domain of the angiopoietin receptor TIE2, which was previously identified as the causative gene for inherited mucocutaneous VM (VMCM).49 However, the mutations differ from the inherited ones, as the most common one (L914F) has never been identified as an inherited mutation and vice versa, the most common inherited mutation (R849W) has never been identified as a sporadic one, suggesting that sporadic mutations probably cause lethality in germline and that inherited one have weaker effects that require additional changes.49
VMCM is caused by amino acid substitutions in the TIE2 gene. These gain-of-function mutations induce phosphorylation of the receptor.50,51 GVM is genetically linked to a locus on 1p21, and caused by loss-of-function mutations in the glomulin gene.43 Currently, there is no correlation between the position of a mutation in the glomulin gene and the characteristics of the disorder, such as the number of glomus cells in the lesion, the extent of the lesion, or the number of lesions. Moreover, for the same mutation, the expressivity is variable from patient to patient.52 So far, a mutation in glomulin has been found in almost all GVM families tested, demonstrating locus homogeneity (Brouillard et al, unpublished).48,52 Among the 30 different mutations found, eight are detected in several families, making efficient genetic diagnosis possible in more than 75% of patients. Several observations suggest a paradominant mode of inheritance for GVM, instead of a simple autosomal dominant transmission. A somatic double-hit mutation has been identified in one GVM resected from a patient with a known inherited glomulin mutation (Aerts et al, in preparation).43 The combination of an inherited and a somatic mutation in the tissue suggests that a complete loss of glomulin is needed for GVM development.43 This could explain the variation in size and location of GVMs, depending on the time of occurrence of the postzygotic second-hit mutations. The low frequency of extensive lesions (>5 cm) and the high frequency of localized lesions (<5 cm) support this model.42
(Table 172-3; Fig. 172-7; and see eFig. 172-7.1)
VM | VMCM | GVM | BRBN | Maffucci Syndrome | |
|---|---|---|---|---|---|
Location | 50% head and neck | Extremities, mucosa | 70% extremities | Especially palms and soles | Extremities |
Skin color | Normal-bluish-purple | Bluish | Bluish-red-purple | Bluish-dark-blue | Normal-bluish |
Aspect | Flat-raised | Raised dome-shaped | Raised, cobblestone Plaque like, hyperkeratotic | Dome shaped, nipple-like | Exophytic |
Single large | Multiple, small | Multiple | Multiple, small often + 1 large lesion | Multiple, important deformation of hands and feet | |
Palpation | Phleboliths | Compressible | Non compressible Painful | Firm, rubbery, Spongy | Firm |
Associated | Coagulopathy | – | – | Visceral venous malformation, coagulopathy | Multiple enchondromas High risk of cancer |
Radiologic | Phleboliths | Venous channels | No phlebolith | Venous channels | Venous channels Enchondromas |
Tissue involved | Often deep location | Cutis and subcutis | Cutis and subcutis | Often deep component | Deep location |
Histology | Thin-walled venous channels | Thin-walled venous channels | Glomus cells | Thin-walled venous channels | Spindle cells Hemangioendothelioma |
Inheritance | Sporadic | Autosomal dominant | Autosomal dominant | Sporadic | Sporadic |
Cause | 50% somatic TIE2 mutations | Gain of function mutation in TIE2 | Loss of function mutation in Glomulin | ? | ? |
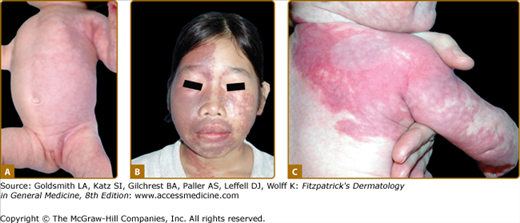
Figure 172-7
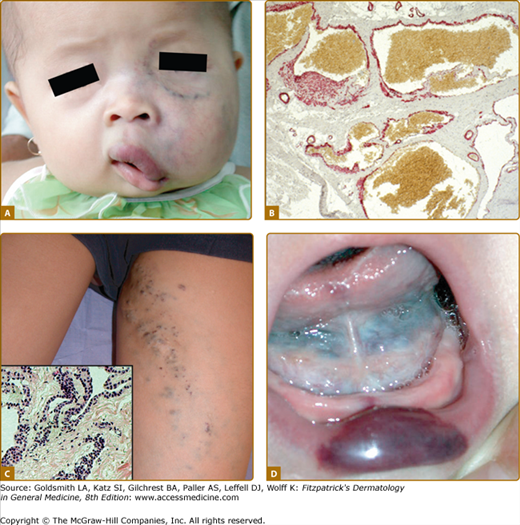
A. Venous anomalies: extensive hemifacial venous malformation (VM) causing soft-tissue hypertrophy and lip deformation in a 6-month-old boy. B. Histologically, VM consists of ectatic venous-like channels with anomalies in mural cells (hematoxylin and eosin). C. Superficial glomuvenous malformation of the inner thigh. Note the presence of mural glomus cells on histology (hematoxylin and eosin). D. Mucosal VM involving the lower lip and the floor of the mouth.
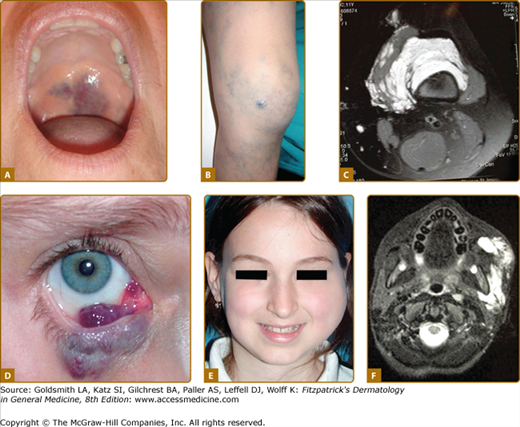
VMs are usually solitary, but can be multifocal, suggesting an inheritable disorder. Cutaneous VMs are congenital light-to-dark bluish lesions. Deep VM has a normal overlying skin color and is often diagnosed only at puberty or later in life with the onset of pain. Cutaneous VMs are detected early because they are usually clinically visible. Size varies from small spongy blebs to large lesions of several centimeters in diameter. VMs can easily be emptied by compression or by positioning in a nondependent position. Skin temperature is normal. There is no thrill or bruit. Depending on size and location, VMs can cause pain, particularly in the morning on awakening, anatomic distortion, and even threaten life because of bleeding, expansion, or obstruction of vital structures. Palpation is not painful unless thrombosis occurs.42 Local thrombosis is responsible for acute pain that lasts for 10 days and causes phleboliths, which can be identified by palpation or by radiography. VM never cause pulmonary embolism. Facial VM is usually unilateral causing asymmetry and progressive deformation and possibly dental malalignment (see Fig. 172-7 and eFig. 172-7.1). Migraine is a common feature, if VM is located in the temporal muscle. Oropharyngeal VM can impair speech, whereas pharyngeal or laryngeal location will compromise the airway and cause snoring and even sleeping apnea. On extremities, VMs often involve muscles and joints (see eFig. 172-7.1). They cause muscle weakness, hypotrophy, and sometimes hypertrophy, resulting in leg length discrepancy, which is less severe than in KTS. Intra-articular bleeding, if the malformation is located in the knee joint, lead to early-onset arthrosis.53 Genital VM often occurs with limb VM and can be responsible for dyspareunia. GI VM is often diagnosed in patients with chronic anemia.
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


