29 Vascular anomalies






Introduction
Vascular anomalies is a newly evolved field that incorporates several surgical and medical specialties. Because these disorders usually involve the skin, the initial consultation is often with a plastic surgeon (or a pediatric dermatologist). Development of this field has been impeded by a lack of standardized terminology. For centuries, it was believed that vascular birthmarks were imprinted on the unborn child by a mother’s emotions or diet. This was reflected in words for brightly colored foods to describe vascular anomalies. Adjectives such as “cherry,” “strawberry,” and “port-wine” have their roots in these traditional beliefs.1 Physicians usually preferred the Latin term naevus maternus for vascular birthmarks, but their understanding was little advanced beyond folklore.
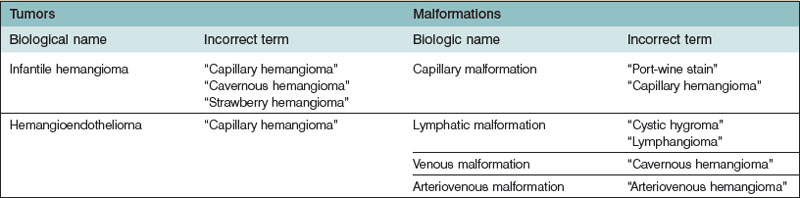
In the 19th century, the first attempt was made to categorize vascular anomalies histologically by Virchow, the father of cellular pathology. Virchow’s angioma simplex became synonymous with “capillary” or “strawberry” hemangioma. His term angioma cavernosum was used indiscriminately for subcutaneous hemangiomas (that regress) and venous malformations (that never regress). Angioma racemosum was modified to racemose (cirsoid) aneurysm or “arteriovenous hemangioma”, referring to an arteriovenous malformation, a vascular lesion that expands over time.2 His student, Wegener, developed a comparable histomorphic subcategorization for “lymphangioma”.3 This nomenclature persisted well into the 20th century. Often the same word was applied to entirely different vascular anomalies. This confusing nosology has been responsible for improper diagnosis, illogical treatment, and misdirected research.
A biologic classification system, introduced in 1982,4 cleared the terminologic confusion that had long obscured the field. This scheme evolved from studies that correlated physical findings, natural history, and cellular features.4 The key to this biologic classification is proper use of the Greek nominative suffix –oma which once meant “swelling” or “tumor”. In modern times -oma denotes a lesion that arises by upregulated cellular growth. There are two major categories of vascular anomalies, tumors and malformations (Table 29.1). Vascular tumors are endothelial neoplasms characterized by increased endothelial turnover (Fig. 29.1). Infantile hemangioma is the most common, a tumor that arises in infants. Other vascular tumors are congenital hemangioma, hemangioendotheliomas, tufted angioma, hemangiopericytomas, angiosarcoma, and pyogenic granuloma. Vascular malformations are the result of abnormal development of vascular elements during embryogenesis (Fig. 29.2). They are designated according to the predominant channel type as: capillary malformation, lymphatic malformation, venous malformation, arteriovenous malformation, and complex forms such as capillary-lymphatic-venous malformation. Malformations with an arterial component are rheologically fast-flow; the remainder are slow-flow.
Table 29.1 Classification of vascular anomalies
| Tumors | Malformations | |
|---|---|---|
| Slow-flow | Fast-flow | |
| Infantile hemangioma (IH) | Capillary malformation (CM) | Arterial malformation (AM) |
| Cutis marmorata telangiectatica congenita (CMTC) | Aneurysm | |
| Telangiectasias | Atresia | |
| Ectasia | ||
| Stenosis | ||
| Congenital hemangioma (CH) | Lymphatic malformation (LM) | Arteriovenous malformation (AVM) |
| Rapidly involuting congenital hemangioma (RICH) | Microcystic | Capillary malformation-arteriovenous malformation (CM-AVM) |
| Macrocystic | ||
| Noninvoluting congenital hemangioma (NICH) | Primary lymphedema | Hereditary hemorrhagic telangiectasia (HHT) |
| PTEN-associated vascular anomaly | ||
| Hemangioendotheliomas | Venous malformation (VM) | Combined malformations |
| Kaposiform hemangio-endothelioma (KHE) | Cerebral cavernous malformation (CCM) | Capillary-arteriovenous malformation (CAVM) |
| Cutaneomucosal venous malformation (CMVM) | Capillary-lymphatic arteriovenous malformation (CLAVM) | |
| Other | Glomuvenous malformation (GVM) | |
| Verrucous hemangioma (VH) | ||
| Pyogenic granuloma (PG) | Combined malformations | |
| Capillary-venous malformation (CVM) | ||
| Capillary-lymphatic malformation (CLM) | ||
| Capillary-lymphatic-venous malformation (CLVM) | ||
| Lymphatic-venous malformation (LVM) | ||
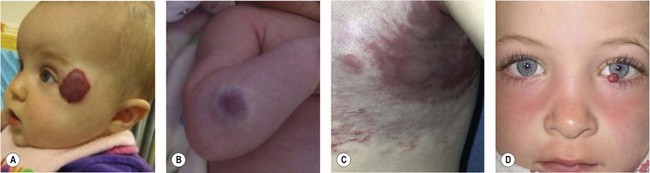
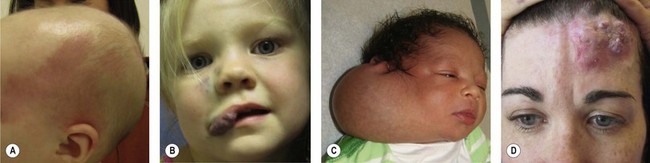
This biologic classification was accepted by the International Society for Vascular Anomalies in 1996.5 Differences that distinguish between hemangiomas and vascular malformations were confirmed by imaging6,7 and by immunohistochemical markers.8,9 It is critical to underscore that vascular malformations, although fundamentally structural disorders, can exhibit endothelial hyperplasia, possibly triggered by clotting, ischemia, embolization, partial resection, or hormonal influences.
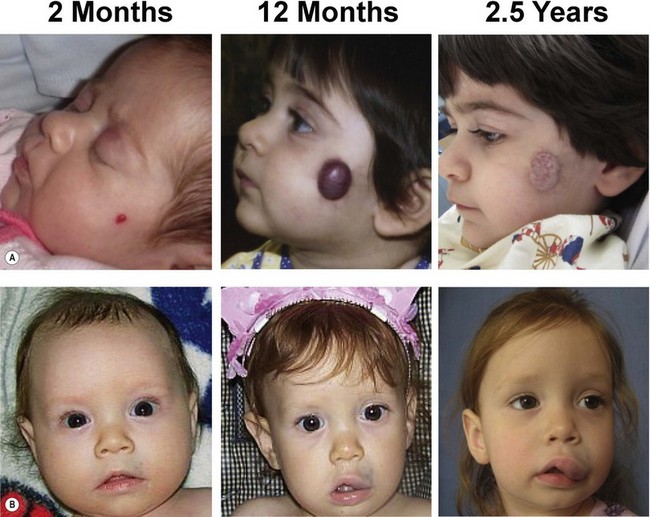
History and physical examination should give diagnostic accuracy of more than 90% in distinguishing between vascular tumors and vascular malformations (Fig. 29.3).10 The most likely error in assigning a clinical diagnosis continues to be an inaccurate, imprecise use of terminology (Table 29.2). Perhaps the most egregious example is “hemangioma”, so often applied generically and indiscriminately to vascular lesions that are entirely different in histology and behavior. There is no such entity as “cavernous hemangioma”. The lesion is either a deep infantile hemangioma or a venous malformation.
Vascular tumors
Infantile hemangioma
Pathogenesis
Infantile hemangioma (IH) is a benign endothelial tumor with a biologic behavior that is unique because it grows rapidly, slowly regresses, and never recurs. There are three stages in its life cycle: the proliferating phase (0–1 year of age), the involuting phase (1–4 years of age), and the involuted phase (after 4 years of age). During the proliferating phase, the histopathologic examination shows clusters of plump endothelial cells with small vascular channels and minimal connective tissue.11,12 During involution, mature blood vessels are formed. Vascular channels enlarge and are lined by flattened endothelial cells. Increased extracellular matrix, multi-laminated basement membranes, and pericytes are deposited around the vessels. After involution, the majority of the IH is replaced with adipocytes and connective tissue. All that remains are thin-walled vessels with multi-laminated basement membranes and larger feeding and draining vessels.13,14
Recent findings suggest that IH arises from vasculogenesis (formation of blood vessels from progenitor cells) rather than angiogenesis (blood vessel formation from pre-existing vasculature).15 Because hemangioma endothelial cells (HemECs) are clonal, a somatic mutation in a precursor cell may be responsible for the tumor.16,17 It is likely that IH begins as an intrinsic (genetic) alteration in a stem cell; its life cycle may be influenced extrinsically, by up or downregulated local angiogenic factors. The precursor cell for IH may be a multipotent hemangioma-derived-stem cell (HemSC), which has been isolated.18 HemSCs express CD90, a mesenchymal cell marker, and can differentiate into multiple cell lineages. They produce human GLUT1-positive microvessels after clonal expansion in immunodeficient mice.18 They differentiate into endothelium and express CD31. HemECs share similarities with placental endothelium,19,20 and it has been postulated that the precursor cell for IH might have embolized from the placenta. Genetic studies, however, have shown that HemECs are derived from the child and not the mother.21,22
Several mechanisms may contribute to the rapid enlargement of IH. Hypoxia may stimulate circulating hemangioma-derived endothelial progenitor cell (HemEPC) recruitment to the growing tumor.15 Increased circulating endothelial progenitor cells have been found in children with IH.23–26 HemEC have been shown to have defective NFAT activity that causes decreased VEGFR-1 expression. Because VEGFR-1 acts as a decoy receptor, more VEFG-A becomes available to bind to VEGFR-2, which stimulates endothelial proliferation.27 Local factors, such as a reduction in anti-angiogenic proteins, also may potentiate tumor growth during the proliferating phase.28
The mechanism for IH involution is unknown. As endothelial proliferation slows, apoptosis increases, and the IH is replaced by fibrofatty tissue. Apoptosis begins before 1 year of age and peaks at 24 months causing a reduction in tumor volume.29 Decreasing circulating maternal estrogens, which are pro-angiogenic, may contribute to involution. Alternatively, increased angiogenesis inhibitors in the epidermis overlying the hemangioma may promote involution.28 The source of adipocytes during involution are HemSCs, which also may differentiate into pericytes.15,18
Clinical features
Infantile hemangioma occurs in approximately 4–5% of Caucasian infants.30 IH is more frequent in premature children and in females (4 : 1).31,32 The tumor is typically single (80%) and involves the head and neck (60%), trunk (25%), or extremity (15%).10 The median age of appearance is 2 weeks; 30–50% are noted at birth as a telangiectatic stain, pale spot, or ecchymotic area.33 IH grows faster than the child during the first 9 months of age (proliferating phase); 80% of its size is achieved by 3.2 (±1.7) months.34 IH is red when it involves the superficial dermis. A lesion beneath the skin may not be appreciated until 3–4 months of age when it has grown large enough to cause a visible mass; the overlying skin may appear bluish. By age 9–12 months, growth of IH reaches a plateau. After 12 months, the tumor begins to regress (involuting phase); the color fades and the lesion flattens. Involution ceases most of children by age 4 years (involuted phase).35 After involution, one-half of children will have: residual telangiectasias, scarring, fibrofatty residuum, redundant skin, or destroyed anatomical structures. There is no strong evidence for Mendelian inheritance of IH;36 however, autosomal dominant transmission has been reported with a locus on 5q.37,38
Multiple hemangiomas
Approximately 20% of infants have more than one IH.33 The term hemangiomatosis designates five or more small (<5 mm) tumors. These children are more likely to have IH of internal organs, although the risk is low. The liver is most commonly affected; the brain, gut, or lung are rarely involved. Ultrasonography should be considered to rule-out hepatic IH.
Hepatic hemangiomas
The liver is the most common extracutaneous site for IH. Some 90% of fast-flow hepatic lesions are IH.39 The differential diagnosis includes arteriovenous malformation, hepatoblastoma, and metastatic neuroblastoma, none of which demonstrate significant shunting on imaging.39 There are three subtypes of hepatic hemangioma: focal, multifocal, or diffuse.40 Although most hepatic IHs are nonproblematic and discovered incidentally, large tumors can cause heart failure, hepatomegaly, anemia, or hypothyroidism. Focal hepatic hemangioma is usually asymptomatic and not associated with cutaneous lesions. There is evidence that solitary hepatic hemangioma is a variant called “rapidly involuting congenital hemangioma (RICH)”.40 Occasionally, this tumor can cause cardiac overload and thrombocytopenia; however, these symptoms resolve as the tumor regresses. Multifocal hepatic IH are often accompanied by cutaneous lesions. Although usually asymptomatic, intrahepatic multifocal lesions can cause high output cardiac failure which is managed by corticosteroid or embolization.40 Diffuse hepatic IH can cause massive hepatomegaly, respiratory compromise, or abdominal compartment syndrome. Infants also are at risk for hypothyroidism and irreversible brain injury because the large tumor volume expresses enough deiodinase to inactivate thyroid hormone.41 Patients require thyroid stimulating hormone monitoring and, if abnormal, intravenous thyroid hormone replacement until the IH begins to regress.
Hemangiomas and structural anomalies
There are uncommon presentations of IH with malformations in the head/neck or lumbosacral regions. PHACE association affects 2.3% of patients with IH, and consists of a plaque-like IH in a regional distribution of the face with at least one of the following anomalies: Posterior fossa brain malformation; Hemangioma; Arterial cerebrovascular anomalies; Coarctation of the aorta and cardiac defects; Eye/Endocrine abnormalities.42 When ventral developmental defects (Sternal clefting or Supraumbilical raphe) are present, an “S” is added (PHACES).42 Of infants, 90% are female and cerebrovascular anomalies are the most common associated finding (72%).43 Because 8% of children with PHACE association have a stroke in infancy, these patients should have an MRI to evaluate the brain and cerebrovasculature.44 Infants are referred for ophthalmologic, endocrine and cardiac evaluation to rule-out these associated anomalies.
Reticular hemangioma is an uncommon variant of IH that most commonly affects the lumbosacral area and lower extremity; 83% of these infants are female.45 Unlike typical IH, reticular tumors are likely to ulcerate; they rarely can cause cardiac overload. Reticular hemangiomas are associated with ventral-caudal malformations (omphalocele, recto-vaginal fistula, vaginal/uterine duplication, solitary/duplex kidney, imperforate anus, tethered cord lipomyelomeningocele).46–50 Ultrasonography (US) is obtained to rule-out associated anomalies in infants less than 4 months of age. MRI is indicated in older infants or when US is equivocal. After involution small veins usually remain; these can be treated by sclerotherapy.
Diagnosis
Most IH are easily diagnosed by history and physical examination. Fast-flow is confirmed using a hand-held Doppler device. By formal ultrasonography, IH appears as a soft-tissue mass with fast-flow, decreased arterial resistance, and increased venous drainage.51 On MRI, the tumor is isointense on T1, hyperintense on T2, and enhances during the proliferating phase.52 Involuting IH exhibits increased lobularity and adipose tissue; the number of vessels and flow is reduced. Rarely, biopsy is indicated if malignancy is suspected or if the diagnosis remains unclear following imaging studies. Tumors or fast-flow lesions that may be confused with IH include: arteriovenous malformation, congenital hemangiomas, cutaneous leukemia (chloroma), hemangioendotheliomas, infantile fibrosarcoma, infantile myofibromatosis, lymphoma, metastatic neuroblastoma, PTEN-associated vascular anomaly, and pyogenic granuloma. If biopsy is needed, positive erythrocyte-type glucose transporter (GLUT 1) immunostaining differentiates IH from other vascular tumors and malformations.9
Nonoperative management
Wound care
During the proliferative phase, at least 16% of lesions ulcerate, the median age is 4 months.53 Superficial IH is prone to ulceration because the skin is damaged by the tumor. In addition, arteriovenous shunting reduces oxygen delivery to the skin, causing ischemia. Consequently, desiccation or minor injury can cause skin breakdown. Tumors located in trauma-prone areas are at greater risk for ulceration; the lips, neck, and anogenital region are the most common locations. To protect against ulceration, IH in these areas should be kept moist with hydrated petroleum during the proliferative phase to minimize desiccation and shearing of the skin. IH in the anogenital area may be further protected by using a petroleum gauze barrier to prevent friction from the diaper.
Topical corticosteroid
Topical corticosteroid is relatively ineffective; especially if IH involves the deep dermis and subcutis.54–56 Ultrapotent agents may be effective for a very superficial IH. Although lightening may occur, if there is deep component it will not be affected. Adverse effects include hypopigmentation, cutaneous atrophy, and even adrenal suppression.57
Intralesional corticosteroid
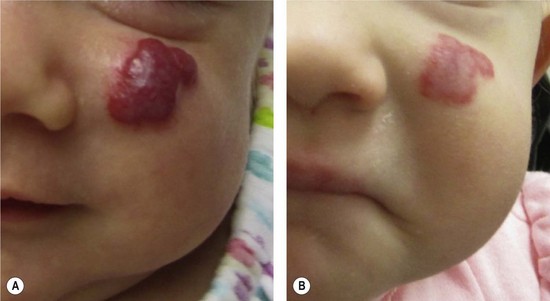
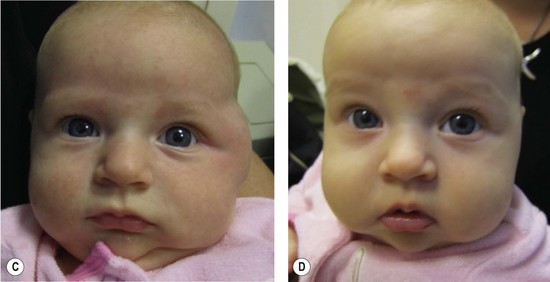
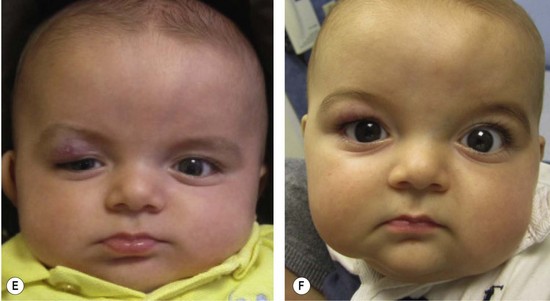
Small, well-localized IHs that obstruct the visual axis or nasal airway, or those at risk for damaging important structures (i.e., eyelid, lip, nose) are best managed by intralesional corticosteroid (Fig. 29.4). Triamcinolone (3 mg/kg) stabilizes the growth of the lesion in at least 95% of patients; 75% of tumors will decrease in size.58 The corticosteroid injection lasts 4–6 weeks, and thus infants may require 2–3 injections during the proliferative phase. Intralesional corticosteroid may cause subcutaneous fat atrophy. Blindness has been reported following injection of deep periorbital hemangioma due to embolic occlusion of the retinal artery.59,60
Systemic pharmacotherapy
Problematic IH that is larger than 3–4 cm in diameter is managed by oral prednisolone or propranolol. Interferon is no longer recommended in children less than 12 months of age because it can cause neurologic sequela, particularly spastic diplegia.61 Vincristine may be considered as a second-line option in the unlikely event that a child has failed or has a contraindication to prednisolone and propranolol.62,63
Oral corticosteroid has been used to treat IH for over 40 years and has proven to be very safe and effective (Fig. 29.4).64–70 Patients are given prednisolone 3 mg/kg/day for one month; the drug is then tapered by 0.5 cc every 2-4 weeks until it is discontinued between 10-12 months of age when the tumor is no longer proliferating. The drug is given once a day in the morning, and infants have monthly outpatient follow-up. Using this protocol, all tumors will stabilize in growth and 88% will become smaller (accelerated regression).70 Treatment response usually is evident within 1 week of therapy by signs of involution: decreased growth rate, fading color, and softening of the lesion. Patients do not require prophylaxis against gastric irritation or prophylactic antibiotics. There are no adverse effects on neurodevelopment.68 Twenty per cent of infants will develop a cushingoid appearance that resolves during tapering of therapy.70 Approximately 12% of infants treated after 3 months of age exhibit decreased gain in height, but return to their pretreatment growth curve by 24 months of age.65
Propranolol has recently proved to be another effective treatment for problematic infantile hemangioma, but its efficacy and safety, compared to corticosteroid has not been studied.71,72 Partial and non response to propranolol has been reported, and potentially serious side effects have occurred: bronchospasm, bradycardia, hypotension, hypoglycemia, seizures, and hyperkalemia.72–78 Propranolol may conceal symptoms of congestive heart failure caused by a large infantile hemangioma, and may further worsen cardiac function.73 Children with asthma/reactive airway disease, blood glucose abnormalities, congenital heart disease, and cerebrovascular malformations (PHACES association) may not be candidates for propranolol.73 Infants receiving propranolol are monitored closely for potential drug toxicity. Patients undergo cardiology consultation, electrocardiogram, echocardiogram, glucose/electrolyte measurements, and frequent blood pressure, heart rate, and respiratory examinations. Children with airway disease, prematurity, or less than 3 months of age undergo inpatient initiation of propranolol.73,77
Laser therapy
There is little, if any, role for pulsed-dye laser treatment for proliferating IH. The laser penetrates only 0.75–1.2 mm into the dermis, and thus only affects the superficial portion of the tumor. Although lightening may occur, the mass is not affected.79,80 These patients have an increased risk of skin atrophy and hypopigmentation.80 The thermal injury delivered by the laser to the ischemic dermis increases the risk of ulceration, pain, bleeding, and scarring.81 Nevertheless, pulsed-dye laser is indicated during the involuted phase to fade residual telangiectasias.
Operative management
Proliferative phase (infancy)
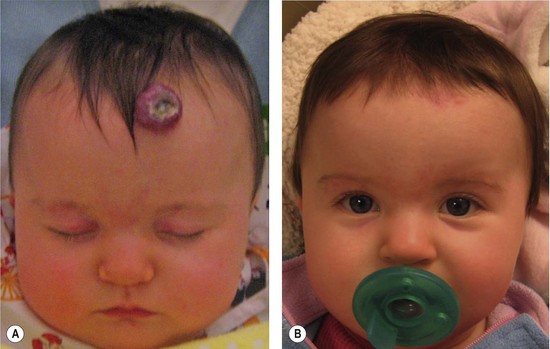
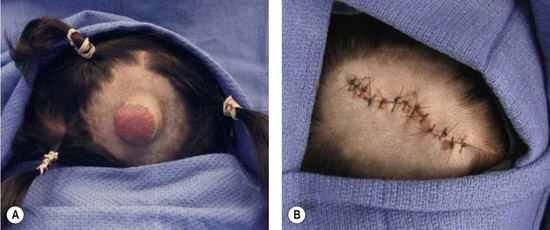
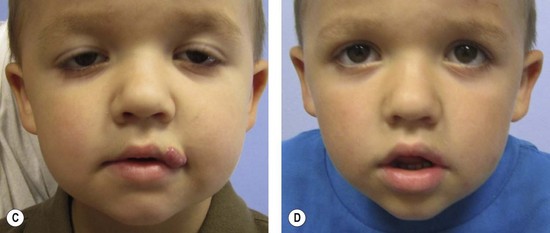
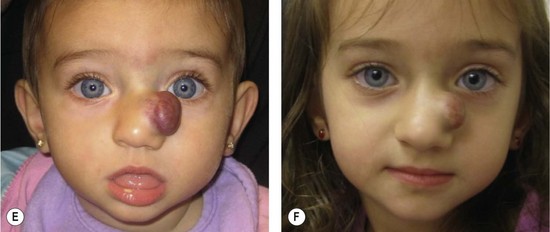
Resection of IH generally is not recommended during the early growth phase. The tumor is highly vascular during this period and there is a risk for blood loss, iatrogenic injury, and an inferior outcome, compared to excising residual tissue after the tumor has regressed.70,82–84 Nevertheless, in experienced surgical hands, there are indications for operative intervention during this phase: (1) failure or contraindication to pharmacotherapy; (2) well-localized tumor in an anatomically favorable area; (3) if resection will be necessary in the future and the scar would be the same (Fig. 29.5).33 Circular lesions located in visible areas, particularly the face, are best removed by circular excision and purse-string closure.85 This technique minimizes the length of the scar as well as distortion of surrounding structures. Lenticular excision of a circular hemangioma results in a scar as long as three times the diameter of the lesion. In comparison, a two-stage circular resection followed by lenticular excision/linear closure 6–12 months later will leave a scar approximately the same length as the diameter of the original lesion.85 Lenticular excision and linear closure is preferred in certain facial locations, such as the lips and eyelids.
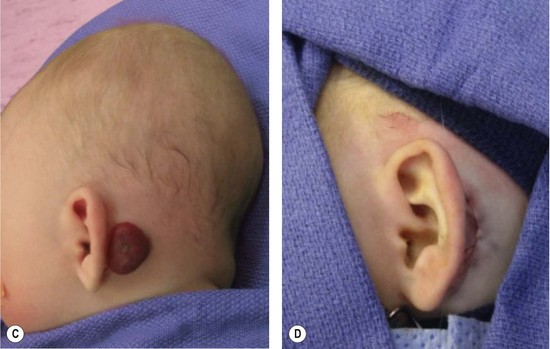
Involuting phase (early childhood)
Resection during involution is much safer because the lesion is less-vascular and smaller. Because the extent of the excision is reduced, the scar is less noticeable. Approximately 50% of IH leave behind fibrofatty tissue or damaged skin after the tumor regresses causing a deformity (Fig. 29.6).33 Sometimes a child requires reconstruction of damaged structures (i.e., nose, ear, lip). Staged or total excision should be considered during this period, rather than waiting for complete involution if: (1) it is clear that the lesion will require resection (e.g., post-ulceration scarring, destroyed structures, expanded skin, significant fibrofatty residuum); (2) the length of the scar would be similar if the procedure was postponed to the involuted phase; (3) the scar is in a favorable location. Another advantage of operative intervention during this period, compared to late childhood, is that reconstruction is underway prior to the child’s development of memory or awareness of a facial difference.
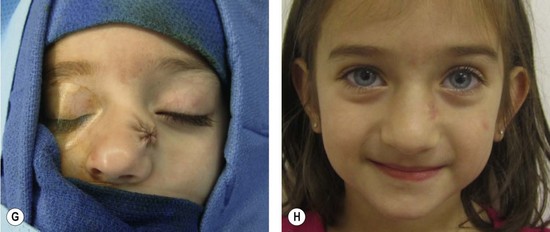
Congenital hemangiomas
Clinical features
There are rare hemangiomas that arise in the fetus, are fully-grown at birth, and do not have postnatal growth.86–88 These congenital hemangiomas are red-violaceous with coarse telangiectasias, central pallor, and a peripheral pale halo. These lesions are more common in the extremities, have an equal sex distribution, and are solitary with an average diameter of 5 cm.86–88 There are two forms: rapidly involuting congenital hemangioma (RICH) and noninvoluting congenital hemangioma (NICH). RICH involutes rapidly after birth and 50% of lesions have completed regression by 7 months of age; the remaining tumors are fully involuted by 14 months.86,88 RICH affects the head or neck (42%), limbs (52%) or trunk (6%). RICH does not leave behind a significant adipose component, unlike common IH. NICH, in contrast, does not regress; it remains unchanged with persistent fast-flow.87 It involves the head or neck (43%), limbs (38%), or trunk (19%).87
Kaposiform hemangioendothelioma
Clinical features
Kaposiform hemangioendothelioma (KHE) is a rare vascular neoplasm that is locally aggressive, but does not metastasize.89–92 Although one-half of lesions are present at birth, KHE may develop during infancy (58%), between age 1–10 years (32%), or after 11 years of age (10%);93 adult-onset is rare.94 KHE has an equal sex distribution, is solitary, and affects the head/neck (40%), trunk (30%), or an extremity (30%).93,95 The tumor is often >5 cm in diameter and appears as a flat, reddish-purple, edematous lesion.91,96 KHE causes a visible deformity as well as pain. Over half the patients have Kasabach–Merritt phenomenon (KMP) (thrombocytopenia <25 000/mm3, petechiae, bleeding).90–92,96 KHE does not exhibit rapid postnatal growth; however, the tumor can expand with the onset of KMP. KHE partially regresses after two years of age, although it usually persists long-term causing chronic pain and stiffness.97 KHE has overlapping clinical and histopathological features with another tumor, tufted angioma, suggesting they are on the same neoplastic spectrum. KMP also may complicate tufted angioma, which has a similar anatomic distribution as KHE, but is more erythematous and plaque-like.98
KHE is diagnosed by history, physical examination, and imaging. MRI is indicated for diagnostic confirmation and to asses the extent of the tumor. MRI shows poorly-defined margins, small vessels, and invasion of adjacent tissues. There is T2-hyperintensity, postgadolinium enhancement, and signal-voids also may be present. Histologically, KHE has infiltrating sheets or nodules of endothelial cells lining capillaries.90,99 Hemosiderin-filled slitlike vascular spaces with red blood cell fragments, as well as dilated lymphatics, are present. Tufted angioma is distinguished from KHE by small tufts of capillaries (“cannonballs”) in the middle to lower third of the dermis.98
Management
Most lesions are extensive, involving multiple tissues, and well beyond the limits of resection. Patients with KMP require systemic treatment to prevent life-threatening complications. Large, asymptomatic tumors without KMP are also managed with pharmacotherapy to minimize fibrosis and subsequent long-term pain and stiffness. Vincristine is first-line therapy, the response rate is 90%.62 KHE does not respond as well to second-line drugs, interferon (50%) or corticosteroid (10%).62,96 Thrombocytopenia is not significantly improved with platelet transfusion because the platelets are trapped in the tumor. Platelet transfusion also worsens swelling and should be avoided unless there is active bleeding or a surgical procedure is planned. By 2 years of age, the tumor usually has undergone partial involution and the platelet count normalizes. There is evidence that KHE never totally regresses.
Pyogenic granuloma
Pyogenic granuloma (PG) is neither “pyogenic” nor “granulomatous”. Some pathologists call it lobular capillary hemangioma.100 PG is a solitary, red papule that grows rapidly on a stalk. It is small, with an average diameter of 6.5 mm; the mean age of onset is 6.7 years.101 The male to female ratio is 2 : 1. PG is commonly complicated by bleeding (64%) and ulceration (36%).101 PG primarily involves the skin (88%), but can also involve mucous membranes (11%). It is distributed on the head or neck (62%), trunk (19%), upper extremity (13%), or lower extremity (5%).101 In the head and neck region, affected sites include: cheek (29%), oral cavity (14%), scalp (11%), forehead (10%), eyelid (9%), or lips (9%).101
Once established, PG rarely spontaneously heals. PGs require intervention to control likely ulceration and bleeding. Numerous methods have been described: curettage, shave excision, laser therapy, and excision.101,102 Because the lesion extends into the reticular dermis, it may be out of the reach of the pulse-dye laser, cautery or shave excision. Consequently, these modalities have a recurrence rate of 43.5%.101 Full-thickness excision is more definitive treatment.
Vascular malformations
Capillary malformation
Pathogenesis
Capillary malformation (CM) is the modern term for the antiquated “port-wine” stain. Its pathogenesis is not understood. The 19th century “neurovegetative theory” asserted that the primary embryonic defect occurs in the developing autonomic nervous system.1 There are several clinical findings that support this hypothesis. The geographic patterns of CMs are often regional or dermatomal (particularly with branches of the trigeminal nerve), suggesting a relationship to the developing peripheral nervous system. The occasional finding of hyperhidrosis in an area of CM supports such an association. Neuroectoderm is known to contribute to the pericytic and smooth muscle layers of vascular walls.103,104 The neurogenic hypothesis also is supported by the finding of decreased perivascular neural density in CMs and abnormal patterns of innervation of leptomeningeal vessels in Sturge–Weber syndrome.105,106 The cutaneous flush of a CM may, in part, be due to an inability of these vessels to constrict secondary to diminished sympathetic innervation.
Clinical features
CMs have an equal gender distribution; the birth prevalence is 0.3%.107 The cutaneous discoloration is usually, but not always, evident at birth because the stain may be hidden by the erythema of neonatal skin. CM often causes psychological concerns as the pink color of childhood darkens and the skin thickens, sometimes with raised fibrovascular cobblestoning. Facial CMs often occur in a dermatomal distribution; 45% are restricted to one of the three trigeminal dermatomes.108 Conversely, 55% of facial CMs are noted to overlap sensory dermatomes, crossing the midline or occurring bilaterally. The mucous membranes often are contiguously involved. Pyogenic granuloma may develop in CM, causing ulceration and bleeding. CM also can lead to soft-tissue and skeletal overgrowth below the stain. When located on the face, hypertrophy of the lip, cheek, or forehead can occur; the lip is most commonly affected.109 Enlargement of the maxilla or mandible can result in an occlusal cant (vertical maxillary overgrowth) with increased dental show and malocclusion.
An extensive CM in an extremity is often associated with increased circumference and limb length discrepancy. CMs in truncal or extremity distributions rarely demonstrate the evolution of textural and color changes seen in facial CMs. CMs often accompany developmental defects of the central neural axis. An occipital CM, often with an associated hair tuft, can overlie an encephalocele or ectopic meninges. A capillary stain on the posterior thorax can signify an underlying arteriovenous malformation of the spinal cord (Cobb syndrome).110–112 A CM over the cervical or lumbosacral spine is a red flag for occult spinal dysraphism, lipomeningocele, tethered spinal cord, and diastematomyelia.33
Management
In patients with an upper facial CM, as well as V1–V2 distribution, the possibility of Sturge–Weber syndrome should be considered. Pulse-dye laser therapy can improve the appearance of CM by lightening the color; the head and neck region responds better than the extremities.113–115 Outcome also is superior for smaller lesions and those treated at a younger age.116,117 Of the patients, 15% achieve at least 90% lightening, 65% improve 50–90%, and 20% respond poorly.118 Improvement is less in Asian patients with facial CMs; only 13.6% of Asian patients show 50% or more lightening.119 A higher rate of complications, including pigmentary changes and hypertrophic scarring, is reported in individuals with darker skin.119,120 After pulse-dye laser treatment, CM often re-darkens over time.121
Facial CM is best treated with pulse-dye laser early in childhood, before memory or self-awareness begins. Intervention in infancy may achieve superior lightening of the lesion, as well as reduce the risk of subsequent darkening and hypertrophy, compared with photocoagulation in later childhood.117 Infants can be treated with pulse-dye laser while awake (using topical anesthesia), depending on the size and location of the CM. After infancy, it is more difficult to restrain an awake child and general anesthesia is preferred, unless the lesion is small. Adolescents generally tolerate laser treatment while awake, depending on the location and extent of the CM. Multiple treatments, spaced 6 weeks apart, are often required until the CM fails to improve with additional treatments. Some families may elect to wait to treat CM of the trunk or extremities until the child is old enough to make the decision. Similarly, patients may wish to undergo laser therapy only if their lesion darkens and becomes more visible over time.
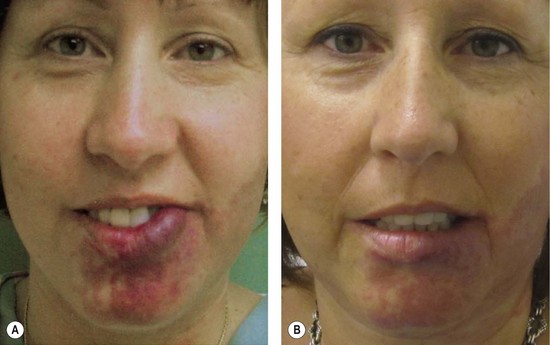
Because overgrowth often is not present at birth and is progressive, most patients do not require contouring, usually labial, until adolescence or adulthood (Fig. 29.7). Cutaneous fibrovascular hypertrophy occurs over many years, requiring intervention in adulthood. Malocclusion can be corrected in adolescence with orthodontic manipulation. If orthodontics is insufficient, an orthognathic procedure is considered when the jaws are completely grown, usually at age 16 years in females and age 18 years in males. Le Fort I osteotomy or bimaxillary procedure may be necessary. Facial asymmetry caused by overgrowth of the zygoma, maxilla, or mandible can be improved by contour burring.
Cutis marmorata telangiectasia congenita
Cutis marmorata telangiectasia congenita (CMTC) manifests as congenital cutaneous marbling, even at normal temperatures, that becomes more pronounced with lower temperatures or with crying.122






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


