Introduction
Vascular anomalies are disorders of the endothelium, which can be subclassified into vascular tumors and vascular malformations, distinguished by proliferating endothelium and quiescent endothelium, respectively ( Table 15.1 ). Vascular anomalies can present in individuals of any age and due to their varied subtypes and morphologies, patients present to clinicians in a wide variety of clinical specialties. These lesions often require significant diagnostic expertise and a multidisciplinary management approach. Arguably, in no area of pediatric medicine is incorrect terminology more pervasive than in the field of vascular anomalies. This is largely due to insufficient teaching on the subject for trainees, the wide variety of terms and labels used in the historic literature and the involvement of many different medical specialties, each with their particular interests and assumptions. Correct designation using the standardized classifications and terminology introduced by the International Society for the Study of Vascular Anomalies (ISSVA) is paramount for appropriate patient management. In the majority of cases, the precise diagnosis can be made from a detailed history and physical examination. In instances where diagnostic ambiguity persists, imaging proves useful, with ultrasound sufficient in most instances. Rarely, is tissue biopsy required.
| Tumors | Malformations | |
|---|---|---|
| Slow Flow | Fast Flow | |
| Infantile hemangioma (IH) | Capillary malformation (CM) | Arterial malformation (AM) |
| Cutis marmorata telangiectatic congenital (CMTC) | Aneurysm | |
| Atresia | ||
| Telangiectasias | Ectasia | |
| Stenosis | ||
| Congenital hemangioma (CH) | Lymphatic malformation (LM) | Arteriovenous malformation (AVM) |
| Rapidly-involuting congenital hemangioma (RICH) | Microcystic | Capillary malformation–arteriovenous malformation (CM–AVM) |
| Noninvoluting congenital hemangioma (NICH) | Macrocystic | |
| Primary lymphedema | Hereditary hemorrhagic telangiectasia (HHT) | |
| PTEN-associated vascular anomaly (PTEN-AVA) | ||
| Hemangioendotheliomas | Venous malformation (VM) | Combined malformations |
| Kaposiform hemangioendothelioma (KHE) | Cerebral cavernous malformation (CCM) | Capillary–arteriovenous malformation (CAVM) |
| Other | Cutaneomucosal venous malformation (CMVM) | Capillary-lymphatic arteriovenous malformation (CLAVM) |
| Glomuvenous malformation (GVM) | ||
| Verrucous hemangioma (VH) | ||
| Pyogenic granuloma (PG) | Combined malformations | |
| Capillary–venous malformation (CVM) | ||
| Capillary–lymphatic malformation (CLM) | ||
| Capillary–lymphatic–venous malformation (CLVM) | ||
| Lymphatic–venous malformation (LVM) | ||
Vascular Tumors
The term vascular tumor encompasses a wide range of pathologies, including common infantile hemangiomas and other rare lesions such as tufted angiomas and kaposiform hemangioendotheliomas. The majority of vascular tumors present in early childhood, have a rapid proliferative growth pattern, and are composed of solid tissue, forming focal soft tissue masses. In contrast, vascular malformations are made up of vascular channels or cysts and are often more diffuse. The most common vascular tumor is the hemangioma . Hemangiomas can be divided into infantile and congenital tumors. Infantile hemangiomas occur in approximately 3%–10% of infants, and in up to 30% of premature infants. These lesions are most common in Caucasian infants, with a lower frequency in infants of African or Asian descent, and are three- to fivefold more common in females than males. In approximately 20% of infants, multiple hemangiomas may be present. Children with multiple infantile hemangiomas, usually five or more, may also have one or more hemangiomas in the liver. These are rarely of any clinical significance. Thus, when five or more infantile hemangiomas are noted, further investigation with abdominal ultrasound is mandatory to exclude the presence of hemangioma in the liver. The term miliary hemangiomatosis describes a rare condition in which a large number of small cutaneous hemangiomas, usually exceeding 20, are apparent soon after birth and recognized to be associated with internal lesions.
Infantile hemangiomas appear to arise from vasculogenesis, and more specifically, from multipotent derived stem cells, which have the potential to differentiate into multiple cell lineages and form human glucose transporter isoform 1 (GLUT1)-positive microvessels in immunodeficient mice. During the proliferative phase, these cells produce vascular endothelial growth factor, an endothelial mitogen. Indeed, GLUT1 is uniquely expressed on the endothelial cells of hemangiomas and provides a valuable marker for differentiation of hemangioma from other vascular tumors and vascular malformations. Hemangiomas have a characteristic evolution, appearing within the first few weeks of life, proliferating for several weeks to months, stabilizing between 6 and 12 months of age, and finally gradually regressing over a period of several years. At birth, the site of the future hemangioma is usually completely normal, with the median age of appearance at 2 weeks. However, in approximately 30% of cases, a nascent lesion may be present, such as an area of telangiectasia or pallor. Rapid growth then ensues, with approximately 80% of tumors attaining their maximal size by 3 months. After 12 months of age, the tumor usually begins the involuting phase. Classically, this stage is characterized by complete resolution of approximately 50% of lesions by 5 years of age, 70% by 7 years of age, and 90% by 9 years of age, which are estimates derived from the study of Bowers et al. However, more recent clinical and histological work has demonstrated that involution is usually complete by age 4 years. Following regression, infantile hemangioma can often leave residual telangiectasia, anetodermic cutaneous excess, fibrofatty residuum, scarring, and, rarely, significant structural deformity ( Fig. 15.1 ).
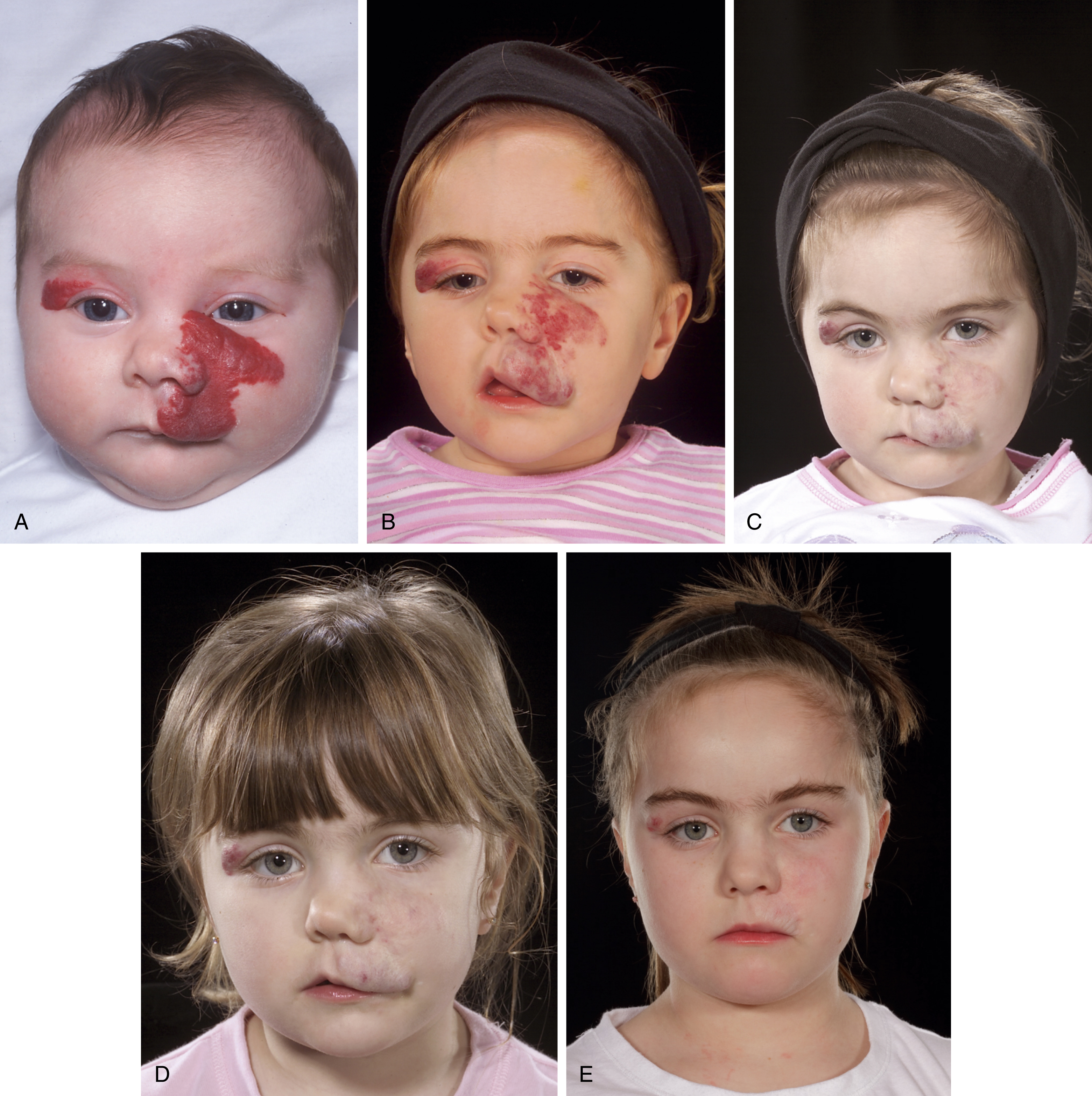
The majority of hemangiomas are small and do not encroach onto anatomically sensitive areas, and as such, are clinically insignificant. These lesions are readily diagnosed in view of their characteristic appearance, including distinctive cherry-red color. Occasionally, deep-seated tumors, which do not alter the color of overlying skin, are encountered, often requiring ultrasound to confirm the diagnosis. With these hemangiomas, families should simply be reassured that the tumor(s) will resolve with time and the lesions followed only if there is cause for concern. Although infantile hemangiomas predictably undergo spontaneous involution, during their proliferative phase, they occasionally cause disfigurement and serious complications, including airway obstruction (subglottic hemangioma), amblyopia (periorbital or orbital hemangioma), cardiac failure, and/or hypothyroidism (hepatic hemangioma), ulceration and bleeding. In the past, such lesions were treated with corticosteroids or laser therapy. In 2008, Léauté-Labrèze et al published a landmark paper where the unanticipated antiproliferative effect of propranolol on infantile hemangiomas was described. Propranolol has since proven to be highly effective and generally well tolerated, and is now the systemic treatment of choice. The therapeutic action of propranolol is secondary to vasoconstriction, inhibition of angiogenesis, and induction of apoptosis. According to a recently published national protocol, the administration of propranolol requires some infants to be given their first dose in hospital and monitoring their heart rate and blood pressure over 2 hours. Adverse effects include bronchospasm, bradycardia, hypotension, hypoglycemia, seizures, hyperkalemia, and diarrhea. Evaluation with a baseline electrocardiogram and/or echocardiogram may be necessary for some infants. A history of asthma, blood glucose abnormalities, and congenital heart disease are considered contraindications for initiation of propranolol therapy. For small superficial hemangiomas, early use of the topical beta-blocking agent timolol, can be effective. The use of systemic corticosteroids is now reserved only as a second-line treatment, when propranolol is contraindicated. There is no role for vincristine or interferon previously documented in the literature. The use of pulsed dye laser should be reserved for treatment of residual telangiectasia following tumor involution. Surgical excision is commonly performed for tumors that are in the involuting phase. This is considered when it is evident that surgical intervention will be necessary and the resultant esthetic outcome is predicted to be comparable to that anticipated during the involuted phase. Otherwise, resection is delayed until involution is complete. Serial excision or circular excision and purse-string closure can be utilized for surgical resection. Occasionally, excision can be considered in the proliferative phase, if medical management is contraindicated or has failed, or as an emergency procedure, for instance a large hemangioma on the upper eyelid causing complete visual obstruction.
Infantile hemangiomas can also occur in extracutaneous areas, most frequently in the liver. Internal organ involvement is more common in infants with multiple cutaneous hemangiomas. These hypervascular hepatic lesions can lead to hepatomegaly and/or hypothyroidism. High output cardiac failure is more commonly associated with congenital liver hemangiomas.
The presence of a flat segmental facial hemangioma should prompt consideration of the associated diagnosis of PHACES . The term comprises p osterior fossa brain malformation, h emangioma, a rterial cerebrovascular anomalies (most common associated finding), c oarctation of the aorta and cardiac defects, e ye/endocrine abnormalities and s ternal clefting or supraumbilical raphe. Evaluation of the brain and cerebral vasculature with MRI is important, although deciding which infants to image remains unclear. Similarly, segmental hemangiomas in the lumbrosacral region and perineum can be associated with ventral-caudal malformations, including omphalocele, imperforate anus, vaginal, and renal anomalies. Radiological investigation (ultrasound and/or MRI) is essential to exclude any associated abnormalities.
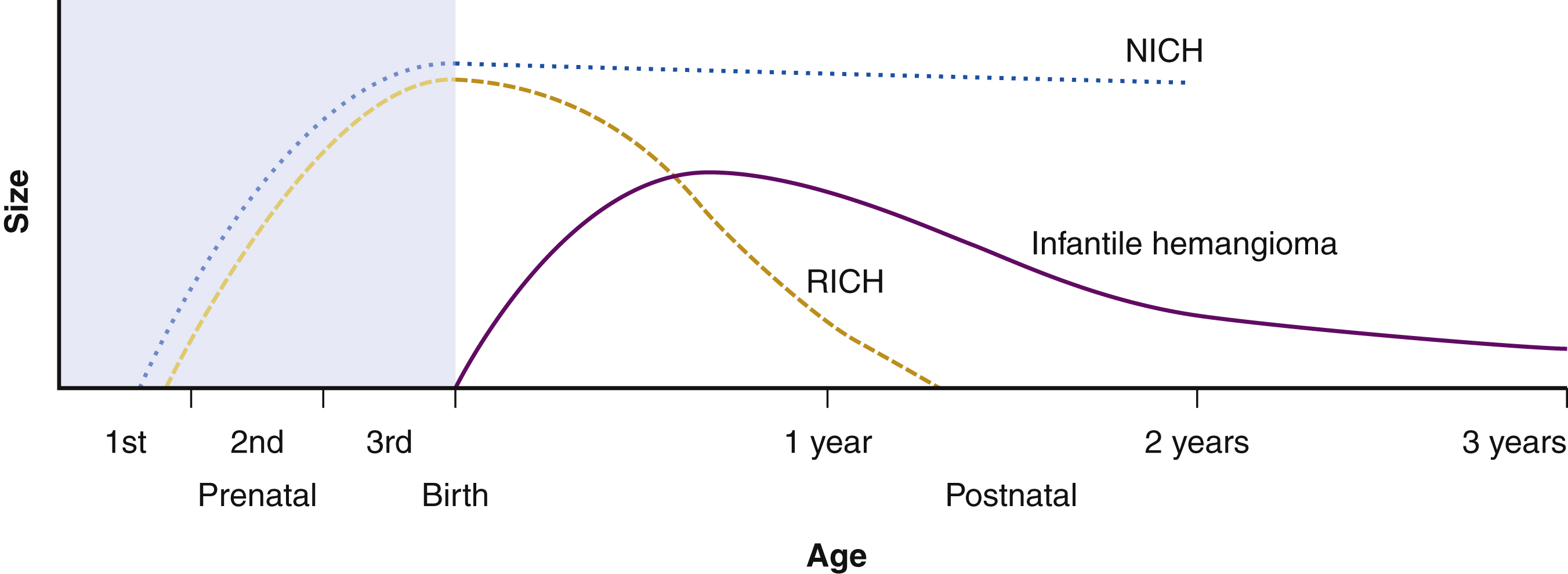
In contrast to infantile hemangiomas, congenital hemangiomas are almost always completely developed at birth and divided into rapidly-involuting congenital hemangiomas (RICH) and noninvoluting congenital hemangiomas (NICH), depending on their pattern of involution. Mulliken and Enjolras have characterized the disparate growth curves for congenital compared with infantile hemangiomas ( Fig. 15.2 ). Aside from the distinctive clinical presentation of these congenital lesions, as well as differing clinical course, negative GLUT1 immunohistochemical staining is an important characteristic to distinguish congenital tumors from their infantile counterparts. There are no distinctive imaging features that reliably differentiate between a RICH and a NICH, with both types appearing as well-defined, hypervascular, relatively homogeneous masses on ultrasound. For some RICH, involution may begin within the first week of life. In most infants with RICH, regression is complete within the first 7–14 months of life, with anetodermic cutaneous excess remaining. Interestingly, in a small proportion of patients with RICH, involution may be incomplete, resulting in a residuum clinically indistinguishable from NICH. A typical NICH forms a flat violaceous vascular plaque with coarse surface telangiectasia bordered by a blue-white rim. Unlike their infantile counterparts, congenital hemangiomas rarely require intervention in infancy. Given the clinical course of RICH, observation is the mainstay of treatment. Occasionally, very large RICH may cause high output heart failure due to their remarkable hypervascularity, in which case embolization may be considered to dampen the lesion’s arterial supply until natural regression occurs. Acute cardiac failure is particularly common with hepatic RICH. Aside from their clinical behavior, hepatic RICH are typically unifocal, and are therefore readily distinguished from hepatic infantile hemangiomas, which are multifocal and usually clinically silent. Large RICH may also result in localized deformity requiring surgical intervention in later childhood. More specifically, these lesions may cause fat atrophy, resulting in contour deformity (in contradistinction to infantile hemangiomas, which often leave a fibro-fatty residuum) or excess anetodermic skin requiring excision. Moreover, regressed lesions can leave an unsightly bed of large, dysplastic veins, which respond well to sclerotherapy. Some lesions may be difficult to differentiate from other tumors, such as congenital fibrosarcoma or rhabdomyosarcoma, and as such may warrant early biopsy and/or surgical excision. NICH may necessitate excision for esthetic concerns as these lesions never involute.
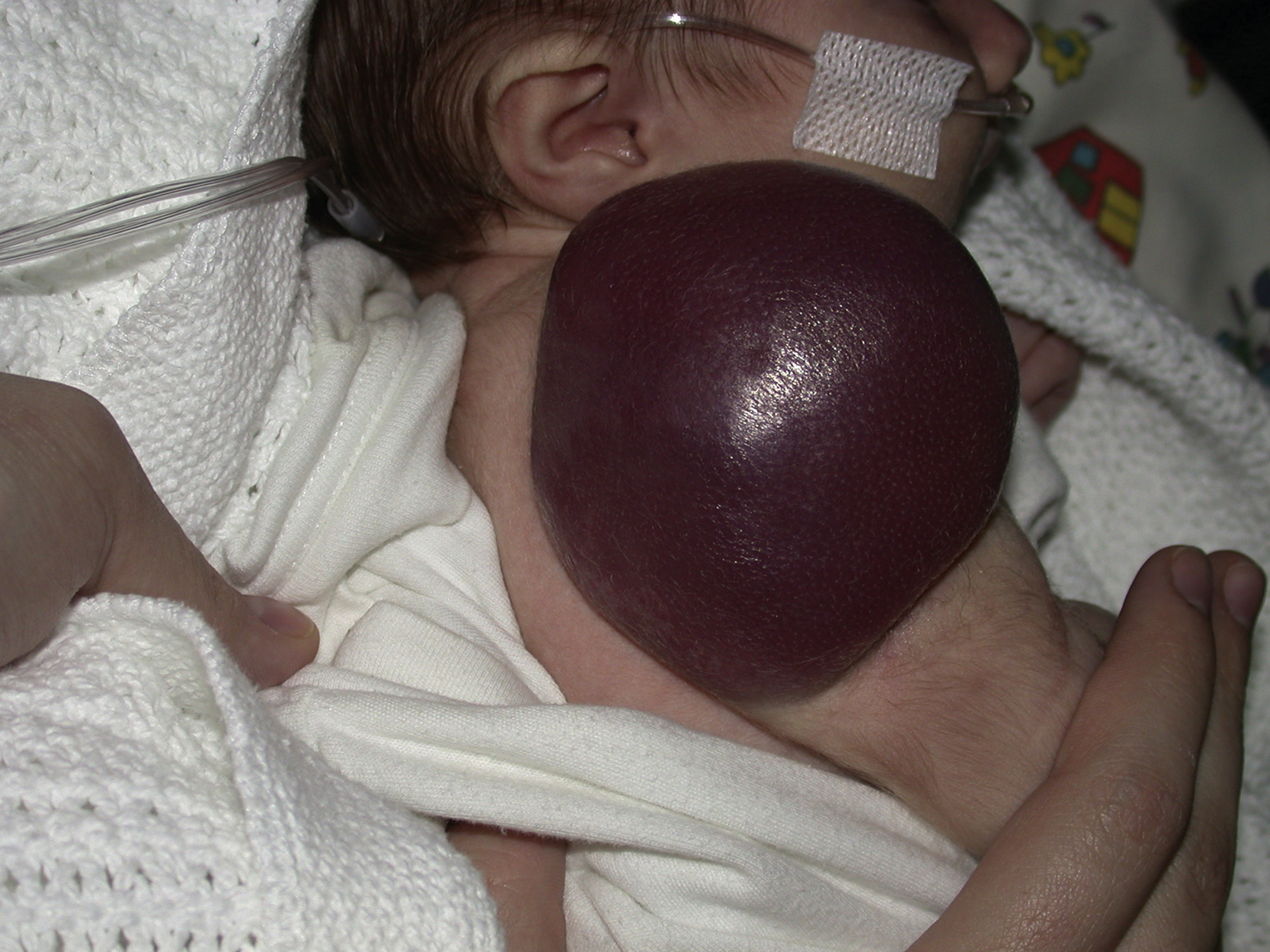
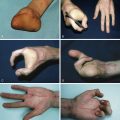
Kaposiform hemangioendotheliomas (KHE) is a locally aggressive solitary vascular tumor, which is usually present at or soon after birth. This tumor often presents as a large violaceous soft tissue swelling ( Fig. 15.3 ). Diagnosis can be confirmed with MRI, which shows a characteristic stranding of the surrounding fat, T2 hyperintensity, poorly defined margins and invasion of adjacent tissues. By 2 years of age, these tumors have usually undergone partial involution but complete involution does not occur. Kaposiform hemangioendothelioma is associated with Kasabach–Meritt phenomenon, characterized by thrombocytopenia <25 ×10 9 /L, petechiae, and bleeding. The other tumor related to this phenomenon is tufted angioma, which is more benign in nature and whose name derives from its histopathologic appearance characterized by tufts of capillaries within the dermis. Tufted angioma and KHE share several clinical and histological features and are thought to be part of the same neoplastic spectrum. The treatment for KHE has traditionally been vincristine, but recent reports of rapamycin (sirolimus), have shown this drug to be highly effective. Platelet transfusion is contraindicated unless there is active bleeding or surgical intervention is planned. While the primary indication for therapeutic treatment is Kasabach–Meritt phenomenon, drug therapy is also considered for large asymptomatic tumors to minimize fibrosis and consequent pain and stiffness.