Varicella and Herpes Zoster: Introduction
|
Epidemiology
Varicella is distributed worldwide, but its age-specific incidence differs in temperate versus tropical climates, and in populations that have received varicella vaccine. In temperate climates in the absence of varicella vaccination, varicella is endemic, with a regularly recurring seasonal prevalence in winter and spring, and periodic epidemics that depend upon the accumulation of susceptible persons. In Europe and North America in the prevaccination era, 90% of cases occurred in children younger than 10 years of age and fewer than 5% in individuals older than the age of 15.1 From 1988 to 1995, there were approximately 11,000 hospitalizations and 100 deaths caused by varicella each year in the United States.2–4 The risk of hospitalization and death was much higher in infants and adults than in children, and most varicella-related deaths occurred in previously healthy people.5 In tropical and semitropical countries, the mean age of varicella is higher and susceptibility among adults to primary VZV infection is significantly greater than in temperate climates. High levels of susceptibility to varicella among adult immigrants from tropical climates are well documented in the US military, where many recruits from Puerto Rico and the Philippines have been seronegative. This is important for hospitals, where susceptible healthcare workers may pose a significant risk of nosocomial varicella.
Widespread use of the varicella vaccine has markedly altered the epidemiology of varicella. In the United States, vaccine coverage rates among susceptible children increased from 0% in 1995, when varicella vaccine was licensed, to 88% in 2004.6 This has resulted in a marked decline in varicella cases and varicella-related hospitalizations. From 1995 through 2000, varicella cases reported to the Centers for Disease Control (CDC) declined by 71%–84%, depending upon surveillance area; by 2005 the incidence of varicella had decreased by 90%, with a comparable decline in varicella-related hospitalizations.7–11 The decline was greatest among children aged 1–4 years, but cases declined in all age groups, including unvaccinated infants and adults, reflecting herd immunity. Varicella-related mortality has also declined substantially after introduction of the varicella vaccine. From 1990 to 1994, mortality from varicella decreased by 66% in all age groups under 50 years, with the greatest reduction (92%) among children 1–4 years of age.4
Varicella is highly contagious. Attack rates of 87% among susceptible siblings in households and nearly 70% among susceptible patients on hospital wards have been reported. More than 95% of cases of varicella are clinically apparent, although occasionally the exanthem may be so sparse and transient as to pass unnoticed. A typical patient is infectious for 1–2 days (rarely, 3–4 days) before the exanthem appears, and for 4 or 5 days thereafter, that is, until the last crop of vesicles has crusted. The immunocompromised patient, who may experience many successive crops of lesions for a week or more, is infectious for a longer period of time. The mean incubation period of varicella is 14 or 15 days, with a range of 10–23 days. It is often prolonged in patients who develop varicella after passive immunization with varicella-zoster immune globulin (VZIG) or zoster immune plasma (ZIP), or after postexposure immunization with live attenuated Oka strain varicella vaccine.11
The major route by which varicella is acquired and transmitted is thought to be the respiratory tract, but infection may also be spread by direct contact. Varicella crusts are not infectious, and the duration of infectivity of droplets containing virus is probably quite limited. Although the infectiousness of patients with varicella is thought to depend largely upon virus shed from the mucous membranes of the upper respiratory tract, VZV has only rarely been cultured from pharyngeal secretions; however, VZV DNA can be detected in the oropharynx of the majority of patients using polymerase chain reaction (PCR)-based assays.12
Natural varicella (i.e., varicella caused by wild-type VZV) generally confers life-long immunity to the disease. Re-exposure to the virus boosts humoral and cell-mediated immune responses, but rarely leads to clinical illness. Most reported second attacks of varicella involve incorrect diagnoses; others may represent cutaneous dissemination in patients with herpes zoster (see below). With severe immunocompromise, reinfections manifested as varicella have been observed. In addition, persons who develop modified varicella (e.g., because they are infected early in infancy in the presence of maternal antibody or have been immunized with live attenuated varicella vaccine) may respond to exogenous exposure by developing a second, usually mild, episode of “breakthrough” varicella.
Herpes zoster occurs sporadically throughout the year without seasonal prevalence. The occurrence of herpes zoster is independent of the prevalence of varicella, and there is no convincing evidence that herpes zoster can be acquired by contact with other persons with varicella or herpes zoster. Rather, the incidence of herpes zoster is determined by factors that influence the host-virus relationship.
One strong risk factor is older age (Fig. 194-1A). The incidence of herpes zoster is 1.5–3.0 per 1,000 person-years in all ages and 7–11 per 1,000 per year in persons over 60 years of age in European and North American studies.13–22 It is estimated that there are more than a million new cases of herpes zoster in the United States each year, more than half of which occur in persons ≥60 years of age, and this number will increase as the population ages.14,17,21,23
Figure 194-1
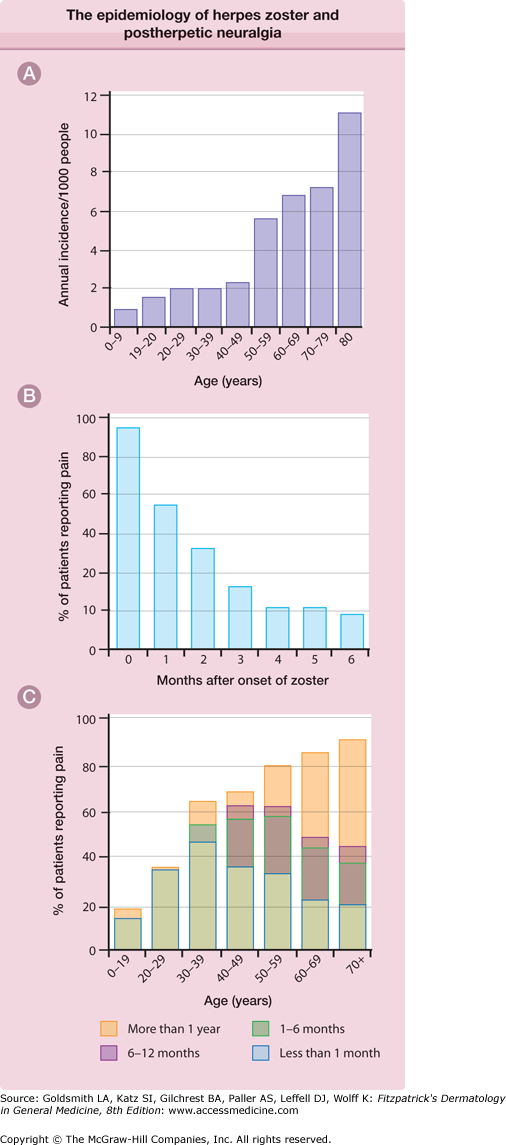
A. The epidemiology of herpes zoster and postherpetic neuralgia. The annual incidence of herpes zoster per 1,000 persons in a general medical practice. B. The percentage of patients with pain persisting after the onset of the herpes zoster rash. These data are from the placebo recipients in one large, double-blind treatment study. C. The proportion of patients with postherpetic neuralgia according to age. (From Kost RG, Straus SE: Postherpetic neuralgia: Pathogenesis, treatment, and prevention. N Engl J Med 335:32, 1996 with permission.)
Another major risk factor is cellular immune dysfunction. Immunosuppressed patients have a 20–100 times greater risk of herpes zoster than immunocompetent individuals of the same age. Immunosuppressive conditions associated with high risk of herpes zoster include HIV infection, bone marrow transplant, leukemia and lymphoma, use of cancer chemotherapy, and use of corticosteroids. Herpes zoster is a prominent and early “opportunistic infection” in persons infected with HIV, in whom it is often the first sign of immune deficiency. Thus, HIV infection should be considered in individuals who develop herpes zoster.
Other factors reported to increase the risk of herpes zoster include female sex,20 physical trauma in the affected dermatome,24 IL-10 gene polymorphisms,25 and white race.19,20 Exposure to children and contact with cases of varicella have been reported to increase levels of VZV-CMI and confer protection against herpes zoster.20,26
Second episodes of herpes zoster are uncommon in immunocompetent persons, and third attacks are very rare. Persons suffering more than one episode may be immunocompromised. Immunocompetent patients suffering multiple episodes of herpes zoster-like disease are likely to be suffering from recurrent zosteriform herpes simplex virus infections.27
Patients with herpes zoster are less contagious than patients with varicella. The rate at which susceptible household contacts develop varicella after exposure to herpes zoster appears to be about one-third of the rate observed following exposure to varicella.14 Virus can be isolated from vesicles and pustules in uncomplicated herpes zoster for up to 7 days after the appearance of the rash, and for much longer periods in immunocompromised individuals. Patients with uncomplicated dermatomal zoster appear to spread the infection by means of direct contact with their lesions. Airborne transmission has also been documented.28 Patients with disseminated herpes zoster may also transmit the infection via aerosols, so that airborne precautions, as well as contact precautions, are required for such patients.
The effect of the marked reduction in the incidence of varicella, due to widespread varicella vaccination of children, on the epidemiology of herpes zoster is unclear. In the long term, the incidence of herpes zoster is likely to decline as the cohorts of children now receiving varicella vaccine become adults; vaccine virus-associated herpes zoster will probably be less frequent and less severe in older adults than wild-type virus-associated herpes zoster because the vaccine virus is highly attenuated. In the short term, the incidence of herpes zoster could increase because a decline in the incidence of varicella will reduce the adult population’s exposure to VZV thereby reducing immune boosting, hastening the age-related decline in immunity to VZV, and thus increasing the age-specific risk of herpes zoster. However, recent studies of herpes zoster in populations with high rates of varicella vaccination have shown little or no increase in the incidence of herpes zoster.8,15,29,30
Etiology and Pathogenesis
VZV is a member of the herpesvirus family.31 Other members pathogenic for humans include herpes simplex viruses type 1 (HSV-1) and type 2 (HSV-2); cytomegalovirus (CMV); Epstein–Barr virus (EBV); human herpesvirus-6 (HHV-6) and human herpesvirus-7 (HHV-7), which cause roseola; and Kaposi’s sarcoma-associated herpesvirus, also called human herpesvirus type 8. All herpesviruses are morphologically indistinguishable and share a number of properties, including the capacity to establish latent infections that persist for life.
The VZV genome encodes about 70 unique genes, most of which have DNA sequence and functional homology to genes of the other herpesviruses.32 Immediate early (IE) gene products regulate VZV replication. Early gene products, such as the virus-specific thymidine kinase and the viral DNA polymerase, support viral replication. Late genes encode virus structural proteins that serve as targets for neutralizing antibodies and cellular immune responses.
There is only one VZV serotype. However, there are multiple VZV genotypes that display geographic segregation and recombination, and minor variations in their nucleotide sequences allow one to distinguish wild type from vaccine virus strains, and to “fingerprint” viruses isolated from individual patients.32,33
Entry of VZV is through the mucosa of the upper respiratory tract and oropharynx. Initial multiplication occurs at this portal of entry, where VZV infects tonsillar T cells, which disseminate virus via the blood and lymphatics (the primary viremia). Infected T cells carry virus to the reticuloendothelial system, the major site of virus replication during the remainder of the incubation period, and to the skin, where innate immune responses delay VZV replication and rash formation.32,34–36
The incubating infection is partially contained by innate host defenses [e.g., interferon, natural killer (NK) cells] and by developing VZV-specific immune responses. In most individuals, virus replication eventually overwhelms these developing host defenses, so that about 2 weeks after infection, a much larger (secondary) viremia and associated symptoms and lesions occur. Skin lesions appear in successive crops, reflecting a cyclic viremia, which in the normal host is terminated after about 3 days by VZV-specific humoral and cellular immune responses. Virus circulates in mononuclear leukocytes, primarily lymphocytes. Even in uncomplicated varicella, the secondary viremia results in the subclinical infection of many organs in addition to the skin. Effective host immune responses terminate viremia and limit the progression of varicella lesions in the skin and other organs. Humeral immunity to VZV protects against varicella. People with detectable serum antibody resulting from wild-type VZV infection do not usually become ill after exogenous exposure. Cell-mediated immunity to VZV also develops during the course of varicella, persists for many years, and protects against severe infections.37,38
During the course of varicella, VZV passes from lesions in the skin and mucosal surfaces into the contiguous endings of sensory nerves and is transported centripetally up the sensory fibers to the sensory ganglia. Infected T cells may also carry virus to sensory ganglia hematogenously. In the ganglia, the virus establishes a latent infection that persists for life. Herpes zoster occurs most often in dermatomes in which the rash of varicella achieves the highest density—those innervated by the first (ophthalmic) division of the trigeminal nerve and by spinal sensory ganglia from T1 to L2.39
Although the latent virus in the ganglia retains its potential for full infectivity, reactivation is sporadic and infrequent, and infectious virus does not appear to be present during latency. The mechanisms involved in reactivation of latent VZV are unclear, but reactivation has been associated with immunosuppression; emotional stress; irradiation of the spinal column; tumor involvement of the cord, dorsal root ganglion, or adjacent structures; local trauma; surgical manipulation of the spine; and frontal sinusitis (as a precipitant of ophthalmic zoster). Most important, though, is the decline in VZV-specific cellular immunity that occurs with increasing age.40
VZV may also reactivate without producing overt disease. The small quantity of viral antigens released during such contained reactivations would be expected to stimulate and sustain host immunity to VZV.14,41
When VZV-specific cellular immunity falls below some critical level, reactivated virus can no longer be contained.14 Virus multiplies and spreads within the ganglion, causing neuronal necrosis and intense inflammation, a process that is often accompanied by severe neuralgia.42,43 Infectious VZV then spreads antidromically down the sensory nerve, causing intense neuritis, and is released from the sensory nerve endings in the skin, where it produces the characteristic cluster of zoster vesicles. Spread of the ganglionic infection proximally along the posterior nerve root to the meninges and cord may result in local leptomeningitis, cerebrospinal fluid pleocytosis, and segmental myelitis. Infection of motor neurons in the anterior horn and inflammation of the anterior nerve root account for the local palsies that may accompany the cutaneous eruption, and extension of infection within the central nervous system (CNS) may result in rare complications of herpes zoster (e.g., meningoencephalitis, transverse myelitis). Viremia also occurs during herpes zoster.44
Pain is a major symptom of herpes zoster. It often precedes and generally accompanies the rash, and it frequently persists after the rash has healed—a complication known as postherpetic neuralgia (PHN). A number of different but overlapping mechanisms appear to be involved in the pathogenesis of pain in herpes zoster and PHN (Fig. 194-2).45,46
Figure 194-2
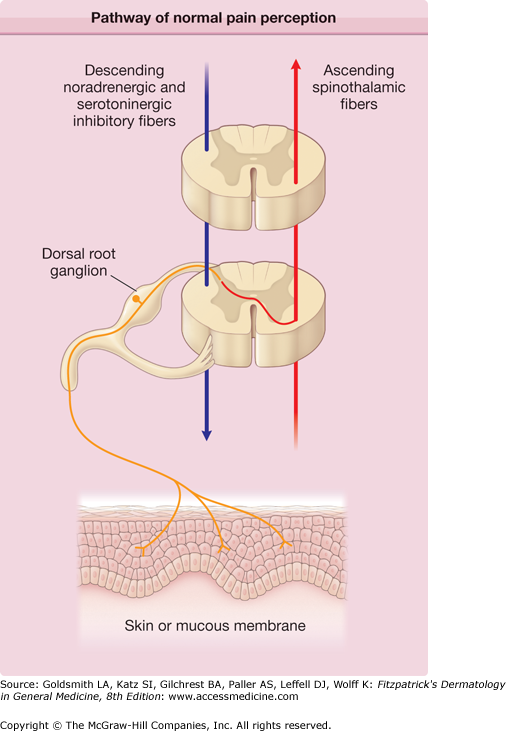
Pathway of normal pain perception. Noxious stimuli activate free nerve endings in the skin to generate signals that are conveyed through unmyelinated C fibers (red) and small Aδ fibers to the neuronal bodies in the segmental dorsal root ganglia, then proximally to the dorsal horn of the spinal cord, where they form synapses with second-order neurons. Spinal cord neurons are subject to powerful descending inhibitory signals from the brain (green), mediated by the biogenic amines serotonin and norepinephrine. Drugs that potentiate the central effects of biogenic amines, such as tricyclic antidepressant drugs, may act by enhancing these descending pathways. Endogenous opiates also contribute to descending inhibitory input. The net result of peripheral afferent input and descending inhibitory input is projected cephalad, joining other ascending fibers in the contralateral spinothalamic tract (orange). Information from the spinothalamic tract is integrated with input from brainstem and cortical areas for the perception of specific aspects of pain as well as more general affective components of pain perception.
Injury to the peripheral nerve and to neurons in the ganglion triggers afferent pain signals. Inflammation in the skin triggers nociceptive signals that further amplify cutaneous pain. The abundant release of excitatory amino acids and neuropeptides induced by the sustained barrage of afferent impulses during the prodrome and acute phase of herpes zoster may cause excitotoxic injury and the loss of inhibitory interneurons in the spinal dorsal horn. Damage to neurons in the spinal cord and ganglion, and to the peripheral nerve, is important in the pathogenesis of PHN. Damaged primary afferent nerves may become spontaneously active and hypersensitive to peripheral stimuli, and also to sympathetic stimulation. Excessive nociceptor activity and ectopic impulse generation may, in turn, sensitize CNS neurons, augmenting and prolonging central responses to innocuous as well as noxious stimuli. Clinically, these mechanisms result in allodynia (pain and/or unpleasant sensations elicited by stimuli that are normally not painful, e.g., light touch) with little or no sensory loss.
The anatomic and functional changes responsible for PHN appear to be established early in the course of herpes zoster. This would explain the correlation of initial pain severity and the presence of prodromal pain with the subsequent development of PHN, and the failure of antiviral therapy to fully prevent PHN (see below).
Clinical Findings
In young children, prodromal symptoms are uncommon. In older children and adults, the rash is often preceded by 2–3 days of fever, chills, malaise, headache, anorexia, severe backache, and, in some patients, sore throat and dry cough.
In unvaccinated persons, the rash begins on the face and scalp and spreads rapidly to the trunk, with relative sparing of the extremities (Fig. 194-3). New lesions appear in successive crops, but their distribution remains central. The rash tends to be denser in the small of the back and between the shoulder blades than on the scapulae and buttocks and more profuse on the medial than on the lateral aspects of the limbs. It is not uncommon to have a few lesions on the palms and soles, and vesicles often appear earlier and in larger numbers in areas of inflammation, such as diaper rash or sunburn.
Figure 194-3
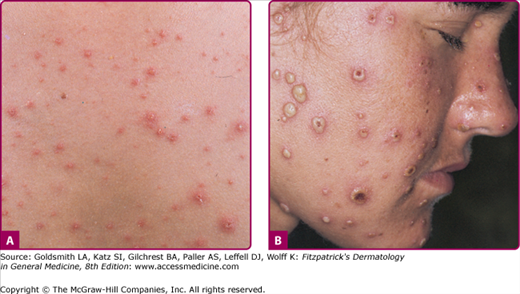
Varicella. A. A full spectrum of lesions—that is, erythematous papules, vesicles (“dewdrops on rose petals”), crusts, and erosions at sites of excoriation—is seen in a child with a typical case of varicella. B. A wider range of lesions, including many large pustules, is seen in a 21-year-old female who was febrile as well as “toxic” and had varicella pneumonitis.
A striking feature of varicella lesions is their rapid progression, over as little as 12 hours, from rose-colored macules to papules, vesicles (Fig. 194-3A), pustules, and crusts. The typical vesicle of varicella is 2–3 mm in diameter and elliptical, with its long axis parallel to the folds of the skin. The early vesicle is superficial and thin-walled, and it is surrounded by an irregular area of erythema, which gives the lesions the appearance of a “dewdrop on a rose petal.” The vesicular fluid soon becomes cloudy with the influx of inflammatory cells, which convert the vesicle to a pustule (Fig. 194-3B). The lesion then dries, beginning in the center, first producing an umbilicated pustule and then a crust. Crusts fall off spontaneously in 1–3 weeks, leaving shallow pink depressions that gradually disappear. Scarring is rare unless the lesions were traumatized by the patient or superinfected with bacteria. Healing lesions may leave hypopigmented spots that persist for weeks to months; if scars occur they are depressed, pox-like.
Vesicles also develop in the mucous membranes of the mouth, nose, pharynx, larynx, trachea, gastrointestinal tract, urinary tract, and vagina. These mucosal vesicles rupture so rapidly that the vesicular stage may be missed. Instead, one sees shallow ulcers 2–3 mm in diameter.
A distinctive feature of varicella is the simultaneous presence, in any one area of the skin, of lesions in all stages of development. Careful prospective studies have shown that the average number of lesions in healthy children ranges from 250 to 500; secondary cases resulting from household exposure are more severe than primary cases resulting from exposure at school, presumably because more intense and prolonged exposure at home results in a higher virus inoculum.
Fever usually persists as long as new lesions continue to appear, and its height is generally proportional to the severity of the rash. It may be absent in mild cases or rise to 40.5°C (105°F) in severe cases with extensive rash. Prolonged fever or recurrence of fever after defervescence may signify a secondary bacterial infection or another complication. The most distressing symptom is pruritus, which is usually present throughout the vesicular stage.
The varicella vaccine alters the natural history of the rash. A small percentage of vaccinees develop “breakthrough” varicella after exposure to people with active VZV infections. The usual breakthrough rash is predominately maculopapular with fewer lesions (i.e., less than 60) and fewer vesicles than the rash of natural varicella. The incidence and severity of fever is also less than that in natural varicella.47
Pain and paresthesia in the involved dermatome often precede the eruption by several days and vary from superficial itching, tingling, or burning to severe, deep, boring, or lancinating pain. The pain may be constant or intermittent and it is often accompanied by tenderness and hyperesthesia of the skin in the involved dermatome. The preeruptive pain of herpes zoster may simulate pleurisy, myocardial infarction, duodenal ulcer, cholecystitis, biliary or renal colic, appendicitis, prolapsed intervertebral disk, or early glaucoma, and this may lead to serious misdiagnosis and misdirected interventions. Prodromal pain is uncommon in immunocompetent persons under 30 years of age, but it occurs in the majority of persons with herpes zoster over the age of 60 years. A few patients experience acute segmental neuralgia without ever developing a cutaneous eruption—a condition known as zoster sine herpete.48
The most distinctive feature of herpes zoster is the localization and distribution of the rash, which is nearly always unilateral and is generally limited to the area of skin innervated by a single sensory ganglion (Fig. 194-4A). The area supplied by the trigeminal nerve, particularly the ophthalmic division, and the trunk from T3 to L2 are most frequently affected; the thoracic region alone accounts for more than half of all reported cases, and lesions rarely occur distal to the elbows or knees.14,18,39
Figure 194-4
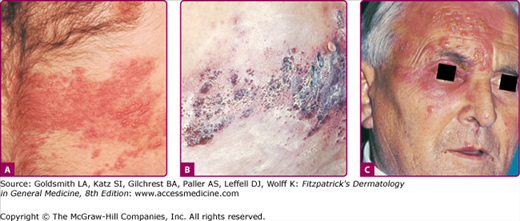
Herpes zoster. A. Early involvement of a thoracic dermatome with erythema within the dermatome and areas of grouped vesicle formation. B. Later involvement with crusted sites on the back, where the eruption first appeared, and many confluent hemorrhagic vesicles and bullae on the lateral chest wall, where the eruption appeared more recently; some vesicles are also seen outside the involved dermatome, representing hematogenous dissemination, a not uncommon occurrence. C. Ophthalmic zoster. Note the involvement of the tip of the nose, which frequently signals involvement of the eye.
Although the individual lesions of herpes zoster and varicella are indistinguishable, those of herpes zoster tend to evolve more slowly and usually consist of closely grouped vesicles on an erythematous base, rather than the more discrete, randomly distributed vesicles of varicella. This difference reflects intraneural spread of virus to the skin in herpes zoster, as opposed to viremic spread in varicella. Herpes zoster lesions begin as erythematous macules and papules that often first appear where superficial branches of the affected sensory nerve are given off, for example, the posterior primary division and the lateral and anterior branches of the anterior primary division of spinal nerves.39 Vesicles form within 12–24 hours and evolve into pustules by the third day. These dry and crust in 7–10 days. The crusts generally persist for 2–3 weeks (Fig. 194-4B). In normal individuals, new lesions continue to appear for 1–4 days (occasionally for as long as 7 days). The rash is most severe and lasts longest in older people, and is least severe and of shortest duration in children.
Between 10% and 15% of reported cases of herpes zoster involve the ophthalmic division of the trigeminal nerve (Fig. 194-4C).49 The rash of ophthalmic zoster may extend from the level of the eye to the vertex of the skull, but it terminates sharply at the midline of the forehead. When only the supratrochlear and supraorbital branches are involved, the eye is usually spared. Involvement of the nasociliary branch, which innervates the eye as well as the tip and side of the nose, provides VZV with direct access to intraocular structures. Thus, when ophthalmic zoster involves the tip and the side of the nose, careful attention must be given to the condition of the eye. The eye is involved in 20%–70% of patients with ophthalmic zoster. Corneal sensation is generally impaired and when impairment is severe, it may lead to neurotrophic keratitis and chronic ulceration.
Herpes zoster affecting the second and third divisions of the trigeminal nerve as well as other cranial nerves may produce symptoms and lesions in the mouth (Fig. 194-5), ears, pharynx, or larynx. The so-called Ramsay Hunt syndrome (facial palsy in combination with herpes zoster of the external ear or tympanic membrane, with or without tinnitus, vertigo, and deafness), results from involvement of the facial and auditory nerves.
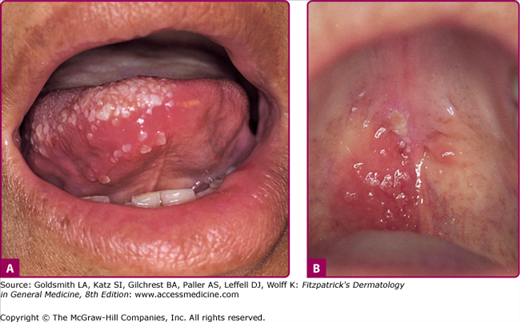
Although the rash is important, pain is the cardinal problem posed by herpes zoster, especially in the elderly. Most patients experience dermatomal pain or discomfort during the acute phase (The first 30 days following rash onset) that ranges from mild to severe. Patients describe their pain or discomfort as burning, deep aching, tingling, itching, or stabbing. For some patients, the pain intensity is so great that words like horrible or excruciating are used to describe the experience. Acute herpes zoster pain is associated with decreased physical functioning, emotional distress, and decreased social functioning.50,51
Except for PHN, most serious complications of herpes zoster occur in immunocompromised persons. These complications include necrosis of skin and scarring (Fig. 194-6) and cutaneous dissemination (Fig. 194-7) with an incidence as high as 25%–50%. Patients with cutaneous dissemination also manifest widespread, often fatal, visceral dissemination, particularly to the lungs, liver, and brain.
Figure 194-6
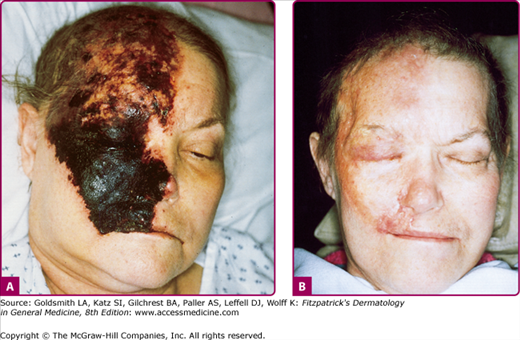
A. Acute, necrotic herpes zoster involving the first and second distributions of the fifth cranial nerve in a woman with lymphoma receiving cytotoxic chemotherapy. B. Dense scar formation and temporal muscle wasting several weeks later. (From Straus SE et al: Varicella-zoster infections: Biology, natural history, treatment and prevention. Ann Intern Med 108:221, 1988 with permission.)
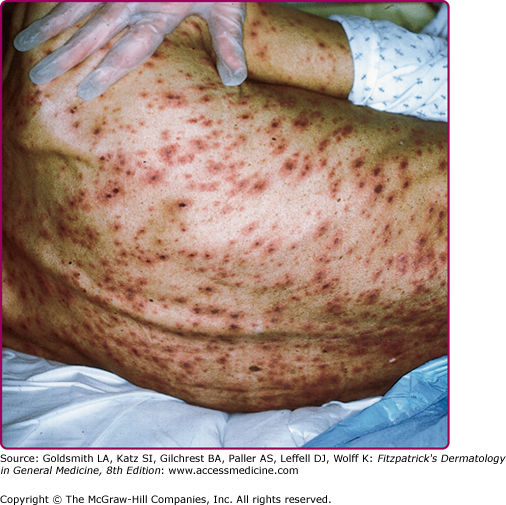
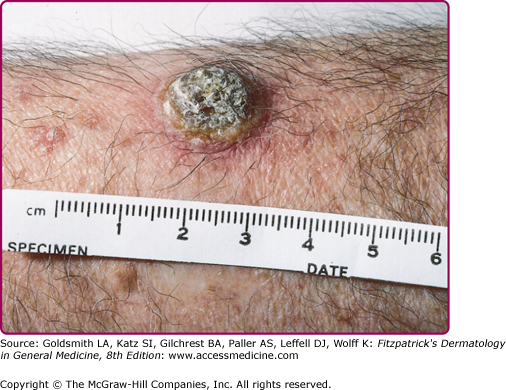
HIV-infected patients are fairly unique in their tendency to suffer multiple recurrences of herpes zoster as their HIV infection progresses; herpes zoster may recur in the same or different dermatomes or in several contiguous or noncontiguous dermatomes. Herpes zoster in patients with AIDS may be severe, with cutaneous and visceral dissemination. Patients with AIDS may also develop chronic verrucous, hyperkeratotic, or ecthymatous cutaneous lesions caused by acyclovir-resistant VZV (Fig. 194-8) (see also Chapter 198).
Differential Diagnosis
Varicella | Herpes Zoster |
|---|---|
Most Likely
| Most Likely
|
Consider
| Consider
|
Always Rule Out
| Always Rule Out
|
Varicella can usually be diagnosed readily on the basis of the appearance and evolution of its characteristic rash (see Fig. 194-3), particularly when there is a history of exposure within the preceding 2–3 weeks.
Disseminated herpes zoster may be mistaken for varicella when there is widespread dissemination of VZV from a small, painless area of herpes zoster or from the affected sensory ganglion in the absence of an obvious dermatomal eruption. This is not infrequent in profoundly immunosuppressed, seropositive persons (Fig. 194-7).
Disseminated HSV infections may resemble varicella; however, there is often an obvious concentration of lesions at and surrounding the site of the primary or recurrent infection (e.g., the mouth or external genitalia) and there may be marked toxicity and encephalitis.
The remaining differential diagnoses of varicelliform rashes are listed in Box 194-1. The character, distribution, and evolution of the lesions, together with a careful epidemiologic history, usually differentiate these diseases from varicella. When any doubt exists, the clinical impression should receive laboratory confirmation.
In the preeruptive stage, the prodromal pain of herpes zoster is often confused with other causes of localized pain. Once the eruption appears, the character and dermatomal location of the rash, coupled with dermatomal pain or other sensory abnormalities, usually makes the diagnosis obvious (Figs. 194-4 and 194-5).
A cluster of vesicles, particularly near the mouth or genitals, may represent herpes zoster, but it may also be recurrent HSV infection.27 Zosteriform herpes simplex is often impossible to distinguish from herpes zoster on clinical grounds. A history of multiple recurrences at the same site is common in herpes simplex but does not occur in herpes zoster in the absence of profound and clinically obvious immune deficiency.
Box 194-1 lists other considerations in the differential diagnosis of herpes zoster.
Laboratory Diagnosis
The lesions of varicella and herpes zoster are indistinguishable by histopathology (Fig. 194-9). The presence of multinucleated giant cells and epithelial cells containing acidophilic intranuclear inclusion bodies (Fig. 194-9B) distinguishes the cutaneous lesions produced by VZV from all other vesicular eruptions (e.g., those caused by variola and other poxviruses, and by coxsackieviruses and echoviruses) except those produced by HSV. These cells can be demonstrated in Tzanck smears prepared at the bedside; material is scraped from the base of an early vesicle, spread on a glass slide, fixed in acetone or methanol, and stained with hematoxylin-eosin, Giemsa, Papanicolaou, or Paragon multiple stain.
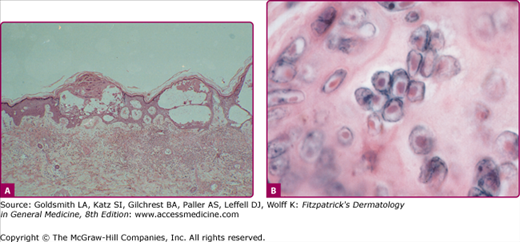
Punch biopsies provide more reliable material for histologic examination than Tzanck smears and facilitate diagnosis in the prevesicular stage and in atypical lesions such as the chronic verrucous lesions produced by acyclovir-resistant VZV in patients with AIDS (Fig. 194-8).
The definitive diagnosis of VZV infection, as well as the differentiation of VZV from HSV, is accomplished by the isolation of virus in cell cultures inoculated with vesicle fluid, blood, cerebrospinal fluid or infected tissue, or by the direct identification of VZV antigens or nucleic acids in these specimens. Virus isolation is the only technique that yields infectious VZV for further analysis, such as determination of its sensitivity to antiviral drugs; however, VZV is extremely labile, and only 30%–60% of cultures from proven cases are generally positive. To maximize virus recovery, specimens should be inoculated into cell culture immediately. It is important to select new vesicles containing clear fluid for aspiration, because the probability of isolating VZV diminishes rapidly as lesions become pustular. VZV is almost never isolated from crusts.
VZV can be isolated and propagated in vitro in monolayer cultures of a variety of human (and certain simian) cells. The cytopathic effects induced by the replicating virus in such cell cultures are characterized by the formation of acidophilic intranuclear inclusion bodies and multinucleated giant cells similar to those seen in the cutaneous lesions of the disease. These changes are indistinguishable from those produced by HSV, but whereas HSV rapidly spreads to infect the remaining cells in the culture, the cytopathic effect of VZV remains focal. Cytopathic effects of VZV are generally not apparent until several days after specimen inoculation. Modifications of the cell culture assay in which vesicle fluid or lesion scrapings are centrifuged onto cells growing on coverslips at the bottom of thin glass-walled “shell” vials followed 24–72 hours later by fixation and staining with fluorescein- or enzyme-labeled monoclonal antibodies to VZV proteins, can confirm the presence of VZV relatively quickly, well before cytopathic effects are evident in conventional cell cultures.52
Immunofluorescent or immunoperoxidase staining of cellular material from fresh vesicles or prevesicular lesions has become the diagnostic method of choice in many centers; it can detect VZV significantly more often and faster than virus culture, even relatively late in the disease when cultures are no longer positive.52 Enzyme immunoassays provide another rapid and sensitive method for antigen detection.
Detection of VZV DNA in clinical specimens following amplifications by PCR provides the greatest assay sensitivity, very high specificity and rapid turnaround time. It has revolutionized the diagnosis of VZV infections, and can distinguish among wild type and Oka vaccine strains of VZV and HSV.52,53
Serologic tests permit the retrospective diagnosis of varicella and herpes zoster when acute and convalescent sera are available for comparison.52 These assays can also identify susceptible individuals who may be candidates for isolation or prophylaxis. The technique most commonly used is a solid-phase enzyme-linked immunosorbent assay (ELISA). However, this assay often lacks sensitivity and specificity, failing to detect antibody in people who are immune and sometimes yielding false-positive results in susceptible individuals. Several more sensitive techniques have been developed to measure humoral responses to VZV. These include an immunofluorescence assay for antibody to VZV-induced membrane antigens [fluorescent antibody to membrane antigen (FAMA)] that reliably distinguishes immune from susceptible adults and a latex agglutination test that is comparable in sensitivity and specificity to FAMA assays, but is much simpler to perform.54
Complications
In the normal child, varicella is rarely complicated. The most common complication is the secondary bacterial infection of skin lesions, usually by Staphylococci or Streptococci, which may produce impetigo, furuncles, cellulitis, erysipelas, and, rarely, gangrene.55 These local infections often lead to scarring and, rarely, to septicemia with metastatic infection of other organs. Bullous lesions may develop when vesicles are superinfected by Staphylococci that produce exfoliative toxins. Invasive group A streptococcal infections are particularly virulent. In the absence of varicella vaccination, up to one-third of varicella is associated with invasive group A streptococcal infections; they usually occur within 2 weeks of the onset of the varicella rash.56 Widespread varicella vaccination appears to have markedly reduced the percentage of invasive group A streptococcal hospitalizations associated with varicella in the United States.57
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


