Differentiation
In tumors, clonality does not always translate into morphologic uniformity. While most sarcomas exhibit only one line of histologic differentiation, a minority may display a strikingly diverse phenotype. This phenomenon not only presents a diagnostic problem but also raises questions about the commitment of tumor cells toward a specific phenotype. Traditionally, the name of mesenchymal tumors is based on their most differentiated pattern; prognosis on the least differentiated. Even though it may be difficult to explain the pathogenesis of divergent differentiation, the variation illustrates that the phenotype of a tumor cell is not set in stone but its direction of differentiation can be influenced (modulated) by a number of factors (1), particularly body location, character of the vascular supply, and signals from surrounding stromal elements.
Modifiers
The usual bewildering array of uniquely pathologic adjectives are applied to variants of the tumors discussed in this chapter, such as myxoid, histiocytoid, epithelioid, angiomatoid, atypical, bizarre, pleomorphic, giant cell, pseudosarcomatous, dedifferentiated, plexiform, storiform, inflammatory, desmoplastic, round cell, clear cell, granular cell, lipomatous, and the like, and their debut is a sure route to acceptance of a case report.
THE BASIC SCIENCE OF FAT RELEVANT TO LIPOCYTIC TUMORS
Obesity, formerly a status symbol of wealth, is now known to be detrimental to health and associated with a wide variety of dermatologic conditions (2). In addition, it is becoming more apparent that obesity as well as neoplasms of adipose tissue are influenced by genetic factors (3).
Adipose tissue can be divided into two types: brown and white. In adults, most fat is the white, univesicular variety. White fat serves as the depot for stored lipid, which is an efficient energy reserve that cushions skin above it and mitigates external trauma. Hematoxylin and eosin (H&E) staining will show only the cell membrane and occasionally the eccentric nucleus with or without vacuoles or pseudoinclusions of cytoplasmic fat (Lochkern, German for “hole in the nucleus”). The red-brown to tan color of brown adipose tissue results from cytochrome pigment in the numerous mitochondria of its finely vacuolated cells. Brown fat has been considered an immature or fetal stage in the development of white fat, but most evidence now substantiates the unique identity of brown fat and its role in basal metabolism and nonshivering thermogenesis. In humans, brown fat is more prevalent in the newborn period than later in life, but it persists throughout adulthood in the neck, axilla, and mediastinum, as well as elsewhere, embedded within the common white fat that, over time, tends to replace it.
Most investigators agree that perivascular cells resembling pericytes and fibroblasts play an important transitional role in the development of benign and malignant lipoblasts. Both white and brown fat cells modulate (Fig. 34-1A) from these primitive fat organ precursors through multivesicular stages easily mistaken for histiocytes, to preadipocytes in which lipid inclusions coalesce and glycogen decreases, and finally to univacuolar adipocytes (4) (Fig. 34-1B) that are variably S100 positive. The multivacuolar stage is observed in various tumors and is likely to reappear with involution following fat injury. In these settings, the cells with foamy cytoplasm are commonly mistaken for histiocytes—for example, the CD68-negative foam cells in the subcutis following biopsy. This is the source of much nosologic confusion, particularly with regard to some of the lesions termed histiocytomas (5).
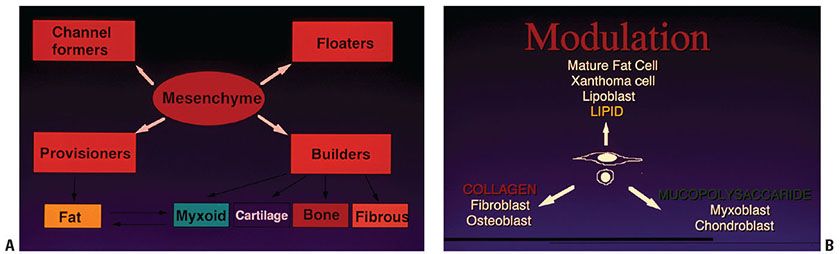
Figure 34-1 Mesenchymal modulation diagrams. A: Uncommitted mesenchyme is the product of differentiation, distinct from ectoderm and endoderm. Further adoption of specific function and corresponding cell structure (phenotype) is modulation, resulting in components of the circulatory system (“channel formers”), lymphohematopoietic cells (“floaters”), synthesizers of structural connective tissue (“builders”), and fat-storing cells (“provisioners”). This normal panoply has its frequent neoplastic parallel in the multiple cell types that can be found in mesenchymal tumors. B: Extracellular matrix products can be collagen-dominant (fibrous or osseous) or mucopolysaccharide-dominant (chondroid or myxoid). A signet-ring fat cell represents the end result of internal product storage rather than export (i.e., accumulation of cytoplasmic fat), a process that begins as multiple small vacuoles resembling a histiocyte. The structure and function of some fully modulated mesenchymal cells can change with altered systemic or local influences in the realms three factors: metabolic (e.g., nutrition, genetically determined enzymatic pathways, endocrine factors, etc.), mechanical (such as trauma, friction, etc.), and/or circulatory (increased arterial flow vs. venous congestion). Initially, in this process, regression leads to spindle or round cells that can be easily mistaken for fibroblasts or lymphocytes. This is followed by cytoplasmic reorganization and acquisition making possible new functions, and more than one modulational potential emerging in mesenchymal tumors as they grow (Figs. 34-8 and 34-9).
Normal fat consists of S100-labeled adipocytes in combination with scattered CD34-positive dendritic cells and small factor XIIIa–positive dendritic cells, more numerous near vessels and within fibrovascular septae. Subsets of them proliferate together in mesenchymal areas of spindle cell lipomas, pleomorphic lipomas, myxoid lipomas, and atypical lipoma/well-differentiated liposarcomas (6).
Fatty tumors of dermatologic interest are common and are generally situated in subcutaneously (7). Aside from nevus lipomatosus, purely cutaneous lipogenic tumors are exceptionally rare and have different specific clinicopathologic features in comparison with more deeply located tumors (8). This fact, as with many other subsets of soft tissue tumors, makes correlation of histopathologic opinions with history and radiologic studies the surest route to accurate diagnoses.
NEVUS LIPOMATOSUS SUPERFICIALIS
Clinical Summary. Nevus lipomatosus superficialis is most often submitted as a solitary small rounded protuberance but can appear clinically as groups of soft, flattened papules or nodules that have smooth or wrinkled surfaces and are skin colored or pale yellow. Characteristically, such multiple lesions are linearly distributed on one hip or buttock (nevus lipomatosus superficialis of Hoffman and Zurhelle) (9) (Fig. 34-2), from where they may overlap onto the adjacent skin of the back, lower torso, pelvis, or upper thigh. Other areas such as the thorax, abdomen, face, or distal extremities are only rarely affected (10–12). The lesions may be present at birth or may begin in infancy with rare presentations of adult onset (10). A rare variant termed nevus angiolipomatosus of Howell (13) refers to the replacement of hypoplastic dermis by pseudotumorous yellow protrusions. It is associated with skeletal and other malformations, developing most commonly during the first two decades of life.
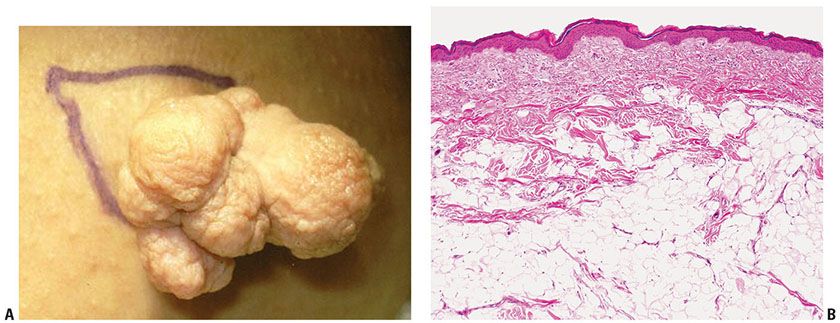
Figure 34-2 Nevus lipomatosus (48-year-old woman, right inner thigh soft mass). A: A 5-cm multinodular exophytic lesion extends from the skin surface. B: Mature signet-ring fat cells emerge in dermal collagen.
Solitary papules or nodules that may not appear until after the fifth decade predilect the trunk and have been diagnosed as solitary nevus lipomatosus superficialis, although the clinical and histologic characteristics are not well delineated from those of acrochordon, soft fibromas (see Chapter 33), polypoid fibrolipomas, or pedunculated lipofibromas (14,15).
Histopathology. Groups and strands of fat cells are embedded among the collagen bundles of the dermis, often as high as the papillary dermis (Fig. 34-2B). The proportion of fatty tissue varies greatly. In examples with only small deposits, the fat cells are apt to be situated in small foci around the subpapillary vessels. In those with relatively large amounts of fat, the fat lobules are irregularly distributed throughout the dermis, and the boundary between the dermis and the hypoderm is ill-defined or lost. The fat cells may all be mature, but in some an occasional small, incompletely lipidized cell may be observed.
Aside from the expansion of fat cells, the dermis may be entirely normal or have a greater density of collagen bundles, number of fibroblasts, and vascularity than normal skin. A slightly undulate or papillomatous epidermis with acanthosis or increased pigmentation may overlie the collections of adipocytes (9).
Pathogenesis. It is generally agreed upon that nevus lipomatosus represents, as the name implies, a nevoid anomaly. The ectopic fat cells in the dermis are derived from the perivascular mesenchymal tissue. This view has found support in electron microscopic studies of nevus lipomatosus superficialis, which in some instances showed, in proximity to capillaries, immature lipocytes containing numerous small lipid droplets and a centrally located nucleus (16). A consistent genetic anomaly has not been defined in these lesions, although there is one report of nevus lipomatosus exhibiting a 2p24 deletion (17,18).
Differential Diagnosis. In focal dermal hypoplasia, fat cells are also found in the dermis, often in close proximity to the epidermis but with concomitant extreme attenuation of collagen (see Chapter 6). Fat cell expansions within long-standing intradermal melanocytic nevi are only exceptionally enough to postulate the association of a nevus lipomatosus with a melanocytic nevus (see Chapter 28). The nomenclature of solitary papules or nodules containing dermal fat becomes rather subjective between solitary nevus lipomatosus, acrochordons with herniated fat, soft fibromas, polypoid fibrolipomas, or pedunculated lipofibromas.
Principles of Management. Nevus lipomatosus superficialis is benign but the lesions may be (and commonly are) surgically removed.
FOLDED SKIN WITH LIPOMATOUS NEVI
Clinical Summary. Multiple, symmetric, deep, gyrate skin folds with underlying nevus lipomatosus or less commonly smooth muscle hamartoma involving the trunk and similar circumferential lesions on extremities since birth are the clinical pathologic features that define the Michelin tire baby syndrome, which was first described in 1969 (19). Often the prominent folds resolve spontaneously. Various associated anomalies of the head and neck as well as systemic features have been described in association with this syndrome, including a round face with hypertelorism, depressed nasal bridge, thin, down-turned vermillion border of the upper lip, and short neck (18,20).
Histopathology. Hypertrophic fat lobules extend close to the epidermis, as in nevus lipomatosus superficialis, and surround adnexal structures (21). In other areas, the dermis is of normal thickness, but excessive amounts of adipose tissue are beneath the dermis and around adnexal structures. Several bands of fibrous tissue penetrate the subcutaneous fat. An increase in smooth muscle fibers of the deeper dermis is reported (22), as well as fragmented elastic fibers (23).
Pathogenesis. Michelin tire baby syndrome has been associated with an autosomal dominant familial syndrome, involving a chromosome 7q inversion in some cases (20). Abnormal elastic fiber formation is thought to be a predisposing factor (23).
Differential Diagnosis. Folded skin has also been reported overlying smooth muscle hamartoma (19,20).
Principles of Management. Most cases improve spontaneously, negating a need for excision or reconstructive surgery (18).
PIEZOGENIC PAPULES
Clinical Summary. Nonneoplastic piezogenic papules present as multiple small papulonodules on the heels (24) or wrist (25), with or without pain, primarily in athletes but also occurring in Ehlers–Danlos syndrome(26).
Pathogenesis. Thickness and septal integrity are both important to the mechanical characteristics of the heel fat pad. In various diseases (e.g., rheumatoid arthritis and diabetes) and aging, the load-carrying ability of the heel pad is impaired (27). Fragmentation and increased width of septal walls is noted in atrophic heel fat pads from patients with peripheral neuropathies (28). Piezogenic papules are due to the herniation of fat into the dermis. In Ehlers–Danlos syndrome, the mechanism of disease is possibly due to structural defects in the connective tissue with resultant poor compartmentalization of the fat (26).
Histopathology. The normally small fat compartments in the lower dermis and subcutis coalesce with loss of fibrous septations (25).
Differential Diagnosis. Bilateral congenital adipose plantar nodules (congenital fibrolipomatous hamartoma) are well-defined lobules of mature fat in the mid and deep dermis, mostly around adnexa (29). They occur in infants along the medial/plantar aspect of the heel rather than the lateral/nonplantar aspect (30).
Principles of Management. Piezogenic papules are primarily an issue for athletes (31) and tend to retract when unburdened. Thus, no treatment is necessary or fruitful.
LIPOMAS
Clinical Summary. Common lipomas consist of mature fat cells throughout (Fig. 34-3) and are the most frequent neoplasms of mesenchyme. Most lipomas (98%) occur as single or multiple subcutaneous growths that are soft, rounded, or lobulated, and movable against the overlying skin; approximately 6% of all cases present as multifocal lesions. Lipomas often display an initial growth phase followed by a period of quiescence. They come to clinical attention only if they reach inordinate size (Fig. 34-3A) or are perceived as blemishes in need of cosmetic removal. The patient commonly presents with an asymptomatic, slowly growing gelatinous or cystic mass. Deep-seated variants of ordinary lipomas tend to be less well circumscribed than their superficial counterparts, often sending out pseudopod-like processes within fascial planes, engulfing aponeuroses, and potentially compressing local structures. Instances of malignant transformation of ordinary lipomas are anecdotal at best.
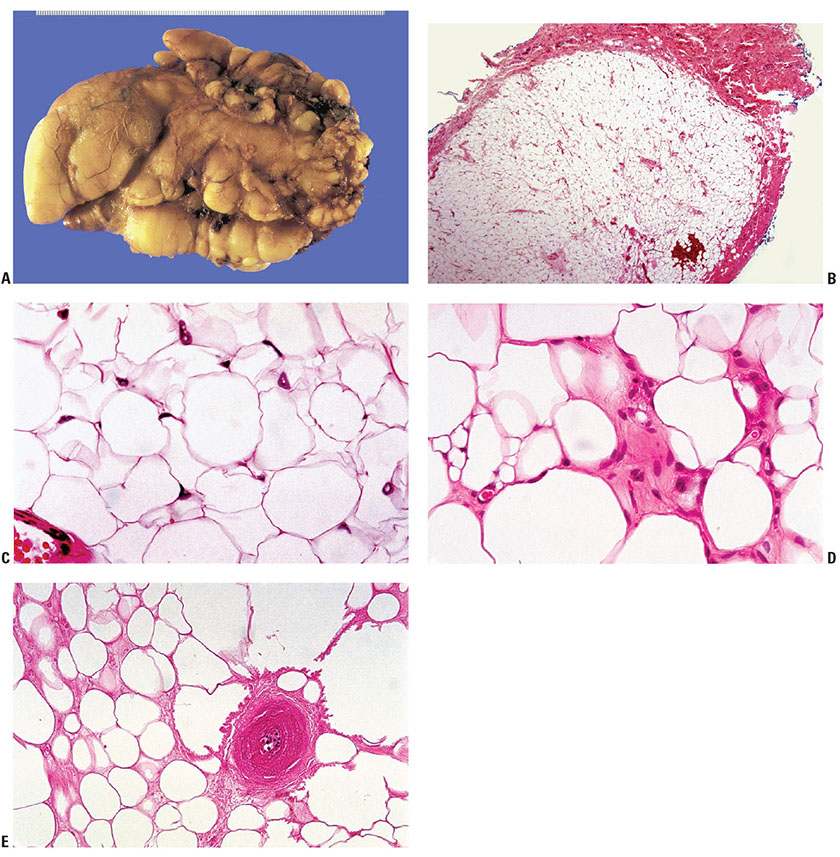
Figure 34-3 Lipoma. A: The pseudopod-like surface irregularities of a lipoma are well shown in this gross example (39-year-old man; groin mass slowly enlarged over 3 years). B: Microscopically, a well-circumscribed collection of mature signet-ring fat cells is bound by a fibrous capsule (51-year-old woman; forehead nodule enlarged over a few months to 12 mm). C: The delicate cell membranes of adipocytes create a “chicken wire” pattern. Nuclei with pseudoinclusions of cytoplasmic fat (Lochkern) as shown here are not to be mistaken for lipoblasts. D: Multivesicular cells (left) and cells display eosinophilic cytoplasm (“hibernoma change,” right). E: Festooned membranous material outlines lipid microcysts around a devitalized vessel in focally necrotic fat of the tumor shown in (A).
Lipomas are subclassified according to anatomic site into dermal, subcutaneous, and subfascial. In addition, they can be directly related to muscle (inter or intra), bone, synovium, or nerve. Some lipomas are characterized by specific clinical presentation and location. The forehead (32) (Fig. 34-3B) is a rather common site specified on dermatopathology requisitions. Perineural lipoma involves the median nerve in 9 out of 10 cases and may be associated with macrodactyly. Lumbosacral lipomas frequently occur in conjunction with spina bifida and intraspinal lipoma. Lipomas of tendon sheath and joint synovium are centered on the named structures. Lipoma arborescens (diffuse articular lipomatosis, synovial lipomatosis, Hoffa disease) is a rare intra-articular lesion of unknown etiology where benign papillomatous intra-articular fatty expansions usually in the knee joint have a tendency for repeated regrowth (33). Intramuscular lipomas, in contrast to intermuscular lipomas, infiltrate muscle and tend to recur unless completely excised (34). Myolipomas of soft tissue are rarely subcutaneous (35).
Many conditions and syndromes are associated with lipomatous tumors or hyperplasias.
In Dercum disease (adiposis dolorosa, lipomatosis dolorosa, morbus Dercum) (36), tender, circumscribed, or diffuse fatty deposits have a predilection for the upper arms, legs, abdomen, and buttocks (37). Postmenopausal women are selectively affected. Paresthesia in the overlying skin is common. Lipomas about the joints cause mechanical arthralgia. The diagnosis, which is often delayed, rests on ultrasonography and, above all, MRI. The portrait painted of Dercum disease is very complicated, and many other disorders are associated with the disease. There are no clear pathologic mechanisms, although it is suspected that there is either a metabolic or autoimmune component. A combination of medications, surgery, and psychiatric care is usually needed, aimed at relieving the pain and restoring a normal appearance.
Multiple or benign symmetric lipomatosis (Madelung disease) is a rare disease of undetermined cause characterized by symmetric deposits of painless, diffuse, subcutaneous adipose tissue on the suboccipital area, cheeks, neck (in “horse collar” distribution), shoulders, and especially on the back and upper trunk (38). It is most common in countries bordering the Mediterranean. The patients are usually middle-aged male alcoholics (39). Although lesions can initially mimic a head and neck malignancy, management is essentially symptomatic, with conservative removals performed as indicated clinically or for cosmesis.
Familial multiple lipomatosis is defined by hundreds of slowly growing subcutaneous (forearm, trunk, and thigh) masses and deep or visceral lipomas that appear in the third decade. This and Dercum disease can be inherited as an autosomal dominant trait, but a conclusive mode of inheritance for the disease process is not known. The HMGA2 gene has been implicated in some studies (40). Children presenting with lipomas need a complete physical examination to look for other signs of phosphatase and tensin homolog (PTEN)–related syndromes (Bannayan–Riley–Ruvalcaba or Cowden syndrome), which are autosomal dominant disorders characterized by germline mutations in the PTEN tumor suppressor gene. This is because these hamartomatous syndromes are associated with tumors of the thyroid, breast, and endometrium in adulthood. Other clinical manifestations of both entities include cutaneous lipomas, facial trichilemmomas, acral keratoses, intestinal polyps, hemangiomas, macrocephaly, and developmental delay (41,42).
Even rarer conditions include diffuse lipomatosis, which features infiltrating masses of mature adipose tissue in part of an extremity or the trunk before age 2 years, possibly with concurrent tuberous sclerosis (43). Nevus psiloliparus (44) is the cutaneous component of subcutaneous scalp lipomas with overlying alopecia in encephalocraniocutaneous lipomatosus (45). Congenital infiltrating lipoma of the face (46) is a rare congenital disorder that occurs in infancy or early childhood in which mature lipocytes extend into adjacent tissue, with associated soft tissue and skeletal hypertrophy, premature dental eruption, and regional macrodontia. Due to its diffuse infiltration and involvement of facial structures, complete surgical excision is often impossible (46). Presenting as a thickened and soft scalp that may present as alopecia, lipedematous scalp is a thickening of the subcutaneous fat (47). Fröhlich syndrome exhibits multiple lipomas, obesity, and sexual infantilism. Lastly, Proteus syndrome features multiple lipomatous lesions, including pelvic lipomatosis and fibroplasia of the feet and hands, skeletal hypertrophy, exostoses and scoliosis, and pigmented skin lesions. This rare overgrowth disorder is generally progressive, with subcutaneous lipomas and cerebriform connective tissue nevi progressively increasing in size, and in most patients, additional lesions developed at new locations (48).
The use of protease inhibitors for HIV-1 can be associated with fat abnormalities including subcutaneous lipomas (49), angiolipomas (50), peripheral lipodystrophy, and benign symmetrical lipomatosis (51).
Histopathology. By definition, lipomas contain mature adipocytes as a principal component. They tend to be surrounded by a thin connective tissue capsule and are composed, often entirely, of normal fat cells that are indistinguishable from the fat cells in the subcutaneous tissue (Fig. 34-3C). Occasional “hibernoma-like” cells or multivesicular cells (Fig. 34-3D) around foci of necrosis should not be misinterpreted as lipoblasts, which differ in having coarse vacuoles and irregular hyperchromatic nuclei. Membranous fat necrosis is a rare occurrence in lipomas of larger size (Fig. 34-3E) and has the typical staining reactions of ceroid pigment. Traumatic or ischemic causation is postulated (52).
Lipomas are also categorized based on histologic details. Intramuscular (infiltrating) lipomas consist of mature fat cells that infiltrate muscle, splaying fibers but showing no nuclear atypia (Fig. 34-4). Be skeptical of this diagnosis on the face where mature fat is normally admixed with facial muscles of expression (Fig. 34-5). Those containing considerable proportions of fibrous connective tissue are called fibrolipomas (Fig. 34-6) or sclerotic lipoma if a prominent collagenous stroma is in a storiform arrangement (53). Substantial basophilic mucopolysaccharide may expand regions of the tumor and the term myxolipoma is recommended (Fig. 34-7).
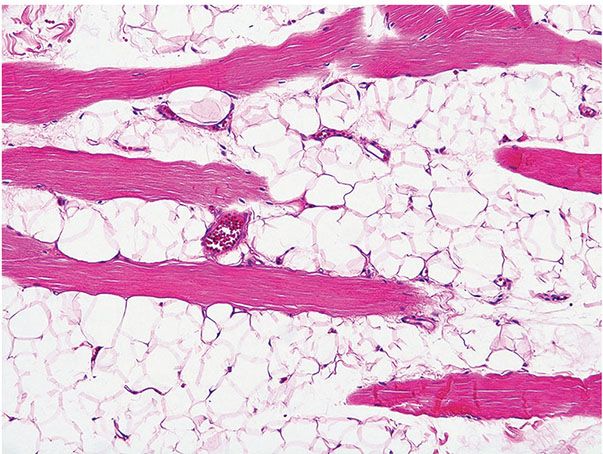
Figure 34-4 Intramuscular (infiltrating) lipoma (from the back of a 57-year-old man). Mature fat cells splay individual skeletal muscle fibers.
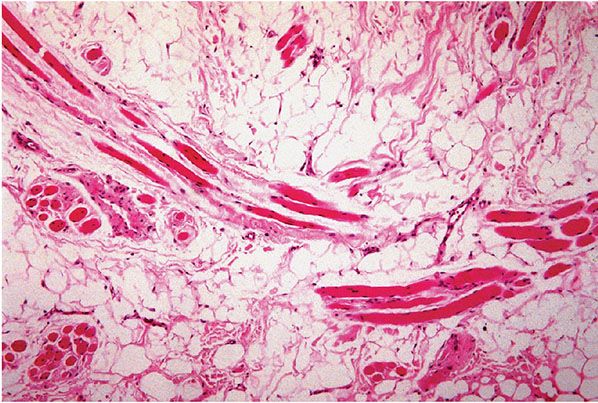
Figure 34-5 Normal subcutaneous tissue, face. Skeletal muscle bundles are normally surrounded by mature fat on the face, and this is not to be mistaken for infiltrating lipoma.
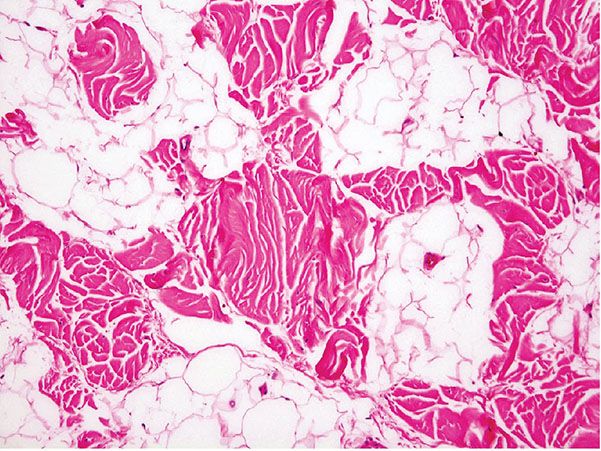
Figure 34-6 Fibrolipoma. Strands of collagenous, fibrous connective tissue are within a tumor composed of mature adipocytes. In other specimens, the nodule may be predominantly fibrous with little fat.
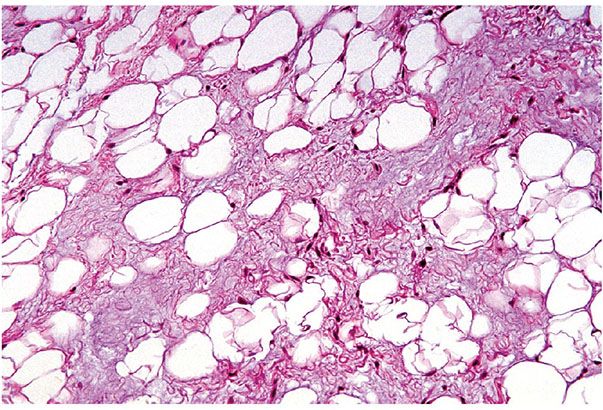
Figure 34-7 Myxolipoma (65-year-old woman; small dorsal hand mass for 7 years, enlarging in the last several months to 2.8 cm). Basophilic myxoid substance between mature adipocytes is sparsely populated by non-atypical stellate- and spindle-shaped cells.
Metaplastic formation of cartilage and bone is only rarely seen in lipomas. Ossifying lipomas, independent of bone tissue and with concurrent cartilage in about half, is rarely reported in the dermatopathologic literature (54). It has the same excellent prognosis as simple lipoma after excision. Focal chondroid (Fig. 34-8) and/or osteoid production (Fig. 34-9) have no significance other than underscoring the modulational potential of functional mesenchymal cells. Mechanical stress, trophic disturbances, character of local vascular flow, the conspicuously frequent contact with periosteum, and still unknown factors may be the causes for the metaplastic transformations (55).
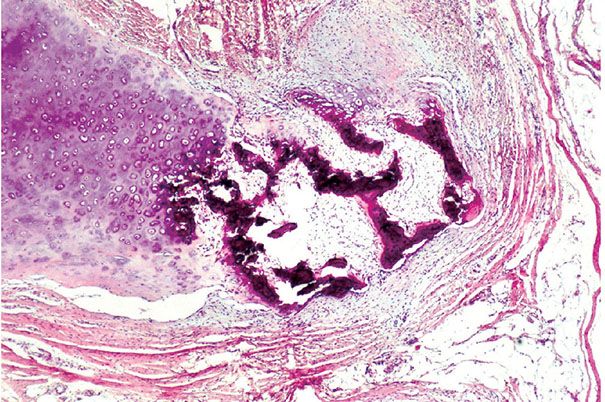
Figure 34-8 Ossifying chondromyxoid lipoma. Within a fatty and myxoid background are areas of chondrification (right) and ossification (center). The contact between the bone and cartilage to the left of center indicates that part of the bone was formed through the enchondral mechanism, analogous to longitudinal bone growth at the growth plate. A portion of the bone has formed directly from the fibrous background (intramembranous ossification). This lesion manifests all three directions of modulation outlined in Figure 34-1.
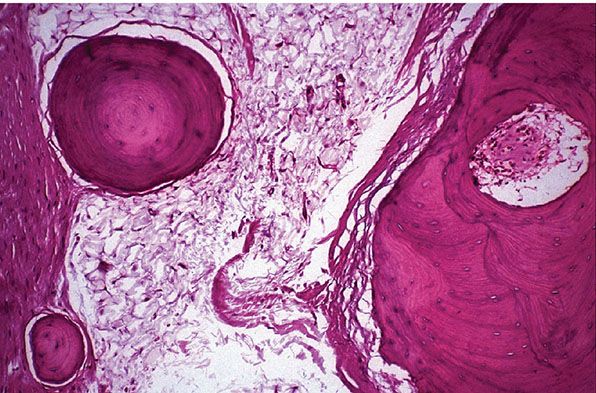
Figure 34-9 Osteolipoma (63-year-old man with “cyst” on forehead for many years). Rounded islands of bone in a fibrofatty background. The largest ossicle (right) is undergoing haversianization.
Myolipomas are comprised of variable amounts of benign smooth muscle and mature adipose tissue. A cutaneous angiomyolipoma is comprised of mature adipose tissue, smooth muscle fascicles, and medium-sized blood vessels (Fig. 34-10), and may show pronounced cellular and nuclear pleomorphism and hyperchromatism of its smooth muscle component, as is common in the renal tumor of the same name (56). The distinctive feature of an adenolipoma (Fig. 34-11) is an admixture of enveloped and distorted sweat glands within the fatty background. The adenolipoma is a dermal or subcutaneous variant of the common lipoma that does not deviate appreciably in presentation or gross appearance but microscopically has inclusions of sweat gland apparatus (8), likely passively enveloped as in “glandular” neurofibroma.
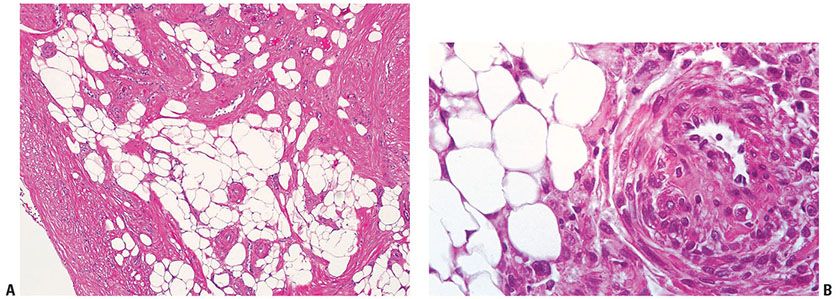
Figure 34-10 Angiomyolipoma. A: The basic components of this tumor are mature adipose tissue and thick-walled, medium-sized vascular channels from which sheets of plump, smooth muscle cells are derived. B: Plump smooth muscle radiates from the wall of a vessel within a fatty background. Moderate pleomorphism of the fatty and smooth muscle components is expected and does not connote malignancy.
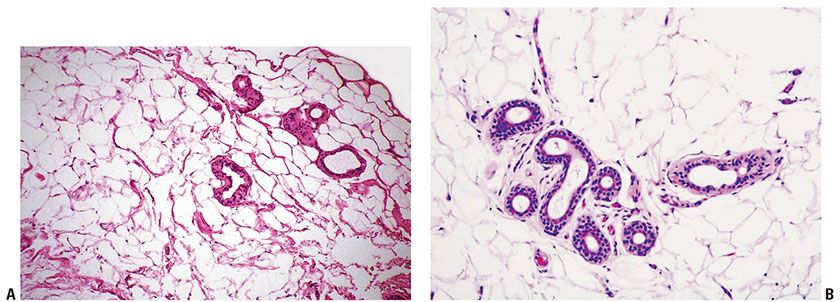
Figure 34-11 Adenolipoma. A: Excess mature fat in this medium-magnification view surrounds sweat gland coils and tubules (52-year-old man; 2-cm left neck nodule). B: Apocrine and sweat gland elements are surrounded by mature fat (62-year-old woman; small axillary mass).
The cutaneous angiomyolipoma, also known as cutaneous angiolipoleiomyoma, is a variant of lipoma seen most often in adult male patients and in an acral location. Like its more common renal counterpart, it is composed of an admixture of blood vessels, smooth muscle, and adipose tissue. While the renal lesions are often seen in the setting of tuberous sclerosis and pulmonary lymphangiomyomatosis, this is not so in the skin, and workup for tuberous sclerosis should not be pursued (57). Smooth muscle cells in cutaneous angiomyolipomas do not react for HMB-45, in contrast to renal and extrarenal angiomyolipoma (8,58). It is proposed that cutaneous angiomyolipomas should be termed angioleiomyoma with fat to avoid confusion with unrelated noncutaneous angiomyolipomas that have HMB-45 positive smooth muscle cells and require investigation for tuberous sclerosis (59).
In cases of syndrome-related lipomas, histologic examination generally reveals adipose tissue indistinguishable from ordinary solitary lipomas. In some cases of adiposis dolorosa, the histologic appearance is that of an angiolipoma, or there may be associated foreign body–type granulomas.
Pathogenesis. Efforts to distinguish true adipocyte neoplasms from lipomatous hamartomas and localized overgrowth of fat have been limited by the lack of reproducible markers for adipose neoplasms. For the present, lipomatous tumors are classified morphologically (Table 34-2), but identification of a (12:16)(q13p11) translocation in myxoid liposarcomas, a translocation in lipomas with breakpoints at 12q14 (60), and the discovery of future chromosomal abnormalities will help in elucidation of pathogenesis and categorization of lipomatous lesions.

Differential Diagnosis. Growth pattern and size may suggest well-differentiated liposarcoma, but superficial (dermal or subcutaneous) liposarcomas are extremely rare. In addition, lipoblasts are found in liposarcoma but are absent in lipoma. Lipomas may have focal or substantial basophilic mucopolysaccharide but the delicate vascular networks of myxoid liposarcoma are not represented. Intramuscular (infiltrating) lipoma lacks the multiple muscle involvement of a pediatric lipoblastoma. Deeply situated myolipomas can be confused with well-differentiated liposarcomas (35).
Principles of Management. Well-circumscribed lipomas are generally easily “shelled out” and only rarely regrow. In cases of generalized lipomatosus, patients may undergo reconstructive surgery. Rarely recurrence may occur, especially when initially incompletely encapsulated or infiltrative. Large, even giant-size lipomas defined as greater than 10 cm diameter in at least one dimension (61), rapid growth, deep location, fixation, pain, and thigh or retroperitoneal location are features that suggest the wisdom of preoperative radiographic characterization and a cautious surgical approach to exclude a more significant lesion before enucleation that could preclude limb-sparing surgery for a more aggressive lesion.
ANGIOLIPOMA
Clinical Summary. Comprising approximately 10% of adipocytic tumors (62), angiolipomas most commonly present as encapsulated subcutaneous lesions in the forearm of young adults, although cases of angiolipoma in the head and neck, including infiltrating tumors, have been reported (63). Clinically, angiolipomas resemble common lipomas, although they have a greater tendency to be multiple and are often tender or painful when pressured or moved (64). Size of these yellow to yellow-red, circumscribed lesions generally ranges from 0.5 to 4 cm.
Histopathology. Unapparent at the gross level, angiolipomas microscopically show sharp encapsulation, numerous small-caliber vascular channels containing characteristic microthrombi, and variable amounts of mature adipose tissue (Fig. 34-12). The ordinary ease of identifying these thrombi on H&E slides can be enhanced with phosphotungstic acid–hematoxylin (PTAH) staining. They are likely artifacts of removal, as they never show organization. The degree of vascularity is quite variable, ranging from only a few small angiomatous foci to lesions with a predominance of dense vascular and stromal tissue (60). The angiomatous foci are composed in part of well-formed, dilated capillaries engorged with erythrocytes. Other capillaries appear tortuous, with poorly formed lumina and proliferated pericytes. Perivascular fibrosis may be prominent. In “cellular” angiolipoma, fat cells are sparse, and small vessels predominate (65). With myxoid stroma, the term angiomyxolipoma (66) can be used. Scattered mast cells are expected. A normal karyotype implies hamartomatous pathogenesis.
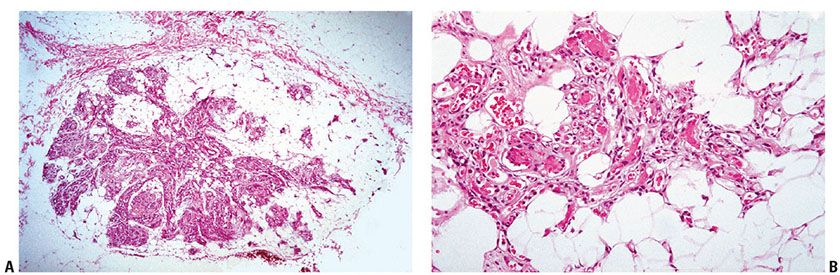
Figure 34-12 Angiolipoma. A: Prominent vessels radiate through a fibrous background into mature fat (incidental finding in a melanoma resection specimen). Prominent vascularity can impersonate a primary vascular neoplasm. B: At higher magnification, fibrin thrombi without organization are in capillary lumina.
Differential Diagnosis. Angiolipoma with prominent perithelial fibrocytes may bring to mind spindle cell lipoma, but spindle cell lipoma lacks the conspicuous capillary channels with fibrin thrombi. Cellular angiolipomas composed almost entirely of vascular channels and prominent spindle cells may have a differential diagnosis that includes Kaposi’s sarcoma or angiosarcoma. Subcutaneous position, encapsulation, septation, small size, diminutive nonatypical endothelial cells, microthrombi, and clinical presentation in healthy individuals help to exclude a malignant diagnosis (67). Also in the differential for angiolipoma is infiltrating angiolipoma (68,69), which is synonymous with intramuscular angioma, a vascular tumor with copious mature fat that is not acknowledged in its name (70). Its vessels are larger and with thicker walls than those in angiolipoma, and it has a significant local regrowth potential.
Nodular–cystic fat necrosis, also called encapsulated angiolipoma, mobile encapsulated lipoma (71), and encapsulated fat necrosis, can be shifted laterally, perhaps by several inches, and cause pain. They are most often in the subcutis of the elbow or hip. The pathogenesis apparently includes ischemic changes secondary to trauma. Nodular–cystic fat necrosis has no permanent attachment to a blood supply. Histologically (Fig. 34-13), the fully developed lesions are thinly encapsulated, rounded nodules with well-preserved outlines of anucleate adipocytes devoid of inflammation or saponification but may calcify. Diffuse formation of pseudomembranes may make diagnosis difficult, leading to consideration of a wide range of potential diagnostic possibilities. This entity should be distinguished histologically from lipoma, angiolipoma, alpha 1-antitrypsin deficiency-associated panniculitis, membranous fat necrosis, and pancreatic fat necrosis (72).

Figure 34-13 Nodular–cystic fat necrosis (“mobile” lipoma). No inflammation assails this encapsulated nodular collection of anucleate (necrotic) adipocytes.
Principles of Management. Management of angiolipoma is similar to that of classic lipomas. When circumscribed, the tumor is easily removed, and when infiltrative, complete surgical excision lowers the chances of recurrence.
SPINDLE CELL LIPOMA
Spindle cell lipoma and pleomorphic lipoma have many overlapping features, and are considered by many to be within a spectrum of the same disease process. This is because they share similar clinical, histologic, and even chromosomal characteristics. For purposes of this chapter, they will be considered separately.
Clinical Summary. A generally solitary tumor, spindle cell lipoma exhibits a predilection for the posterior neck and shoulder girdle region in men in their fifth to seventh decades, presenting as a slowly growing, painless nodule centered in the dermis or subcutis and measuring from 1 to 13 cm (73), but usually less than 2.5 cm when found in the dermis (74). This tumor does not recur or metastasize.
Histopathology. Although the usual deeper lesion is well circumscribed, spindle cell lipoma is seldom encapsulated, and when found in the skin, may have poorly defined borders (74). It is comprised of mature fat cells and uniform, slender spindle cells within a mucinous matrix (75), with a polymorphous appearance as a result of variations in cellularity, collagen content, and the ratio of spindle cells to mature adipocytes. In addition, the cytologic atypia may vary, with progression of atypia from focus to focus (Figs. 34-14A–C). In some areas, the neoplasm consists of a pure spindle cell population, often arranged in thick bundles, without fat cells (Fig. 34-14A). In other areas, the spindle cells intermingle with scattered groups of mature fat cells (Fig. 34-14B). The spindle cells, functioning as fibroblasts, produce varying amounts of collagen. If abundant, the diagnosis is fibrous spindle cell lipoma (76). Numerous mast cells are scattered throughout the tumor (Fig. 34-14C). Some tumors also show a prominent admixture of blood vessels ranging from capillaries to thick-walled vessels containing smooth muscle bundles to prominent sinusoidal channels that divide the tumor into irregular lobules. Irregular branching spaces between villiform, vascularized tumor projections constitute the pseudoangiomatous variant (77). Osseous or cartilaginous metaplasia is rare. About 10% are the “low-fat” and “fat-free” spindle cell lipoma variants easily confused with nerve sheath tumors (78). A number of other variants have been reported, including composite spindle cell lipoma–hibernoma, and composite spindle cell lipoma/pleomorphic lipoma.
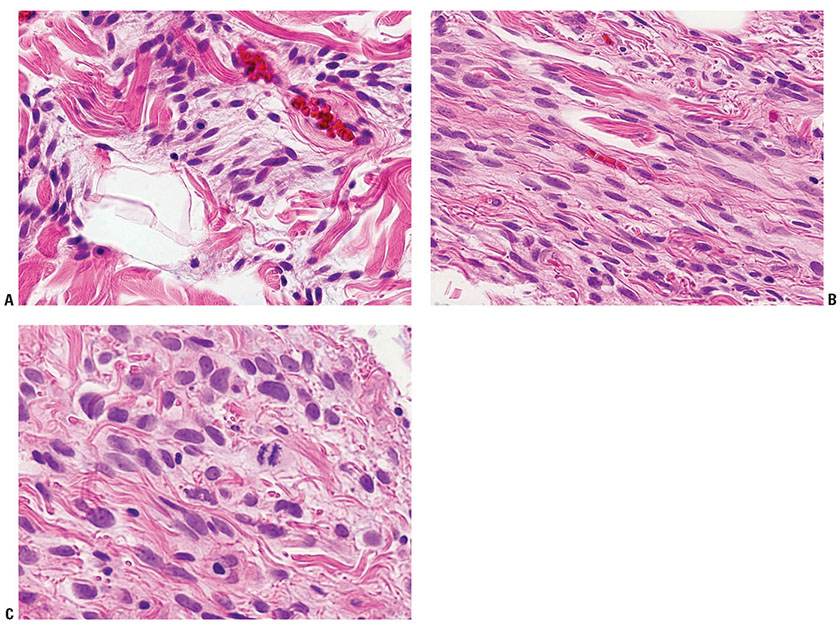
Figure 34-14 Three spindle cell lipoma specimens illustrate the potential range of spindle cell atypia from A to C.
The spindle cells are positive for CD34 and vimentin but not S100. Some factor XIIIa–positive stromal cells are present. Ultrastructurally, they have abundant rough endoplasmic reticulum similar to fibroblasts and non–membrane–bound lipid vacuoles (79). They do not react with MDM1 or CDK4 (80) and may lose reactivity for Rb if there is loss of 13q (81). The cytogenetics parallels pleomorphic lipoma with loss of 16q material and less frequently 13q (82,83).
Differential Diagnosis. Nuclear palisading may be reminiscent of a neural tumor. Myxoid change and hypocellularity may bring myxoma to mind. A prominent hemangiopericytoma-like vascular pattern may be seen or a plexiform vascular pattern that may resemble myxoid liposarcoma. The diagnosis of liposarcoma or fibrosarcoma can usually be excluded without difficulty because of the uniformity of the proliferated spindle cells and the absence of multivacuolated lipoblasts, nuclear atypia, or atypical mitotic figures in spindle cell lipoma (75). In cases with pleomorphic lipoma overlapping features, there may be some nuclear atypia, atypical cells, and atypical mitotic figures; larger univacuolated cells with some atypia, with or without floret giant cells, constitute transitional features to pleomorphic lipoma. In such cases, immunohistochemical reactions for MDM2 and CDK4 are helpful, as they are negative in spindle and pleomorphic lipoma and positive in atypical lipomatous tumors (80), and loss of Rb occurs in spindle cell and pleomorphic lipoma but not in atypical lipomatosus tumors (81). Consider also mammary type and extramammary myofibroblastoma and solitary fibrous tumor (84).
Principles of Management. Spindle cell lipoma/pleomorphic lipoma is benign and can be conservatively excised. Even if incompletely excised, this lesion rarely recurs.
PLEOMORPHIC LIPOMA
Clinical Summary. Like spindle cell lipomas, the great majority of pleomorphic (giant cell) lipomas are solitary tumors of the shoulder girdle and neck in men in the fifth to seventh decade. The rare cutaneous spindle lipomas and pleomorphic lipomas, in contrast to subcutaneous forms, lack circumscription (74), are more common in females, and have a wider anatomic distribution, including the head and neck, shoulder and upper back, trunk, and lower limbs and upper limbs (8,74). The intradermal lesion presents as a soft, slowly growing cutaneous nodule that is usually less than 2.5 cm; the more common subcutaneous forms may be larger (74).
Histopathology. This tumor displays a wide morphologic variation. Although areas of mature fat cells are present, some of them show enlarged, hyperchromatic nuclei. In addition, most adipocytes show marked variation in size, and rarely are entirely absent in the represented fields (85). In the original series of 48 cases from the Armed Forces Institute, approximately half of the tumors contained occasional multivacuolated cells with the appearance of lipoblasts (86). The mature and immature fat cells are situated singly and in groups in a mucinous stroma traversed by dense, ropy, birefringent collagen bundles. Pseudopapillary structures as in the pseudoangiomatous variant of spindle cell lipoma may be seen (87). Characteristic giant cells exhibit, within an eosinophilic cytoplasm, multiple, marginally placed, and often overlapping hyperchromatic nuclei termed floret-type giant cells for their resemblance to small flowers (Fig. 34-15) (86). Rarely, pleomorphic lipoma histology comprises a sharply demarcated nodule within an otherwise ordinary lipoma. Also, small foci of spindle cell lipoma may be encountered in some pleomorphic lipomas. Some pleomorphic lipomas have inflammatory cells, including lymphocytes, plasma cells, mast cells, and occasional histiocytes in a perivascular or diffuse stromal distribution. Mitoses are rare but occur, and when they do may be atypical.
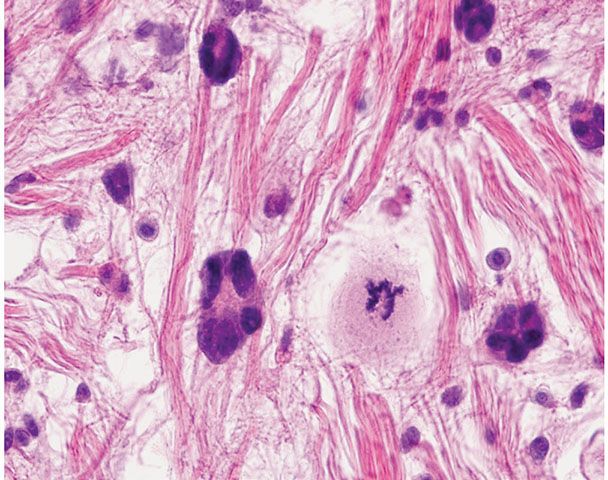
Figure 34-15 Pleomorphic lipoma with local regrowth (59-year-old man; interscapular local regrowth that had regained its original size of 9 cm 8 years after initial surgery). A characteristic floret-type giant cell with multiple peripherally placed nuclei in deeply eosinophilic cytoplasm is near center in ropy collagen beside an atypical mitosis as may occur in pleomorphic lipoma.
Pathogenesis. Like spindle cell lipoma, there is loss of chromosome 16q material (82).
Differential Diagnosis. Pleomorphic lipoma microscopically may resemble a liposarcoma (88). However, it is a slow-growing and well-circumscribed lesion with benign behavior. Despite occasional lipoblast-like cells and atypical mitotic figures, local excision should be curative. No single feature confirms the diagnosis of pleomorphic lipoma and excludes liposarcoma. Only a multivariate analysis leads to the correct diagnosis, and such an analysis should consider the age and sex of the patient, anatomic location, size, epicenter of growth (deep or superficial), degree of local invasion, and histologic appearance. Liposarcomas usually differ from pleomorphic lipomas by their infiltrative growth, greater cellularity, more nuclear atypicality including atypical mitoses, more numerous multivacuolated lipoblasts, prominent necrosis, and absence of thick collagen bundles, although some nuclear atypia and atypical mitotic figures do occur in pleomorphic lipoma. As in spindle cell lipoma, MDM2, CKD4, and Rb immunohistochemical studies can be helpful in this regard. Floret-type giant cells are rarely seen in liposarcomas and then only in small numbers (86).
Principles of Management. Spindle cell lipoma/pleomorphic lipoma is benign and can be conservatively excised. Even if incompletely excised, this lesion rarely recurs.
CHONDROID LIPOMA
Clinical Summary. Chondroid lipoma is an uncommon firm, yellow tumor generally encountered in the proximal lower limb and limb girdles of women (89). These nonpainful masses of weeks’ or years’ duration range from 1.5 to 11.0 cm in diameter, with a median of 4.0 cm. Perhaps half of these well-demarcated, encapsulated, yellow-to-white or gray-tan masses are in the subcutis or involve the superficial fascia of the skeletal muscle (Figs. 34-16A, B). The remainders are more deeply situated, sometimes within muscle. This lesion, almost invariably mistaken for a sarcoma, especially myxoid liposarcoma or myxoid chondrosarcoma, is nonaggressive and has a benign clinical course without local regrowth or metastases on follow-up of the original 12 cases (35).
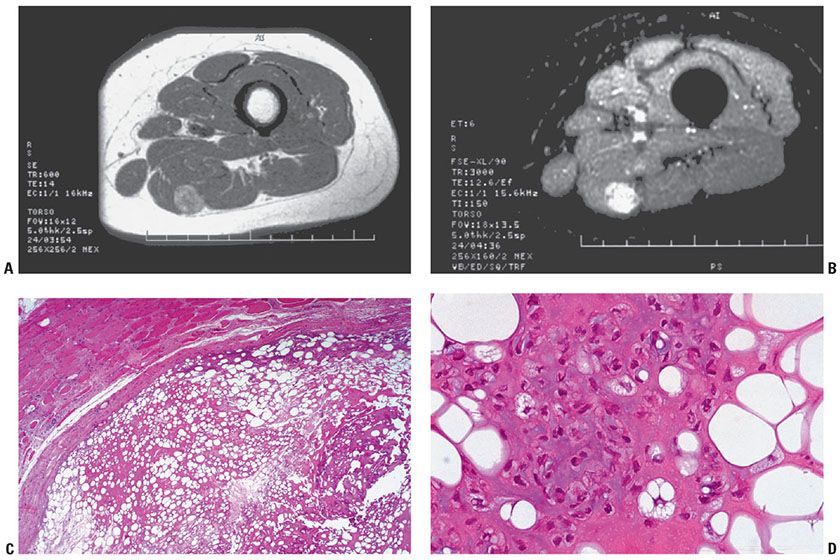
Figure 34-16 Chondroid lipoma (60-year-old woman; 6-month history of posterior leg nodule). A: A T1-weighted MRI of proximal thigh reveals a well-demarcated intramuscular mass with areas of high and low to intermediate signal consistent with fat. B: In a short tau inversion recovery (STIR) image, the foci of high T1 signal suppress, indicating fat. The bulk has high signal consistent with increased water content (myxoid) and cartilage. C: Chondroid lipoma is a lobular, well-circumscribed tumor comprised of strands and nests of rounded or polygonal eosinophilic cells in a stroma of mature fat, myxoid and chondroid material, and partly hyalinized fibrous tissue. D: The eosinophilic tumor cells have lipid vacuoles and grossly simulate lipoblasts. They contain neutral fat and glycogen and stain with Oil Red O and PAS. They are positive for vimentin and S100 protein.
Histopathology. In chondroid lipoma, a variable background of mature adipose tissue is associated with a predominant, partially fibrinous to hyalinized myxoid matrix (Fig. 34-16C). Within this matrix are nests, strands, and sheets of eosinophilic and vacuolated cells, which contain glycogen and fat droplets, resembling brown fat cells, lipoblasts, and chondroblasts (90). The eosinophilic cells frequently contain one or several clear lipid vacuoles and an appearance similar or indistinguishable from lipoblasts. Vacuolated cell forms (hibernoma-like cells) simulate lipoblasts or chondroblasts. These cells lack pleomorphism and mitoses and adopt a lacunar appearance when surrounded by the prominent myxoid or hyaline matrix (Fig. 34-16D). The sulfated stroma is strongly metachromatic with toluidine blue at pH 4.0, and is Alcian blue positive, hyaluronidase resistant, and aldehyde fuchsin positive at pH 1.7.
The characteristic reaction panel is positivity for S100 protein, CD68, and KP1. Chondroid lipoma can be diagnosed by fine-needle aspiration biopsy (91), especially with immunochemistry and special stains performed on the cell block.
Pathogenesis. Electron microscopy reveals that the tumor cells have abundant intracytoplasmic lipid and glycogen as well as numerous pinocytotic vesicles characteristic of adipocytes rather than chondroblasts. This favors the view that chondroid lipoma is a tumor of adipocytes with a hyalinized extracellular matrix that resembles cartilage under light microscopy (92). Another view is that the cells have features of embryonal fat and embryonal cartilage (93). Female predominance raises the possibility of hormonal factors in pathogenesis (94). Recently, the presence of the C11orf95-MLK2 fusion gene, correlating with the t(11;16)(q13;p113) translocation, has been identified in cases of chondroid lipomas by real time polymerase chain reaction (RT-PCR) analysis (90). A three-way translocation between chromosomes 1, 2, and 5, together with an 11;16 translocation with a breakpoint in 11q13, is reported (95).
Differential Diagnosis. The differential diagnosis includes primarily extraskeletal myxoid chondrosarcoma (where myxoid matrix obscures the mature fatty component), soft tissue chondroma (mainly hands and feet), myxoid liposarcoma, hyaline variant of chondroid syringoma, and occasionally myoepithelial tumors. A plexiform, capillary-like vasculature and myxoid matrix are features of myxoid liposarcoma (94).
Principles of Management. This benign tumor can be surgically removed if cosmetically or functionally indicated.
HIBERNOMA
Clinical Summary. The constituent cells of a hibernoma resemble those of brown fat tissue, which is present in the human fetus and newborn infants but gradually diminishes in quantity throughout adult life. Hibernomas may appear in childhood and slowly increase in size but present chiefly in adults, with a peak incidence during the third decade of life, which is an age range considerably younger than those with ordinary (mature) lipoma. This rare tumor arises frequently at the few sites in which brown fat is encountered in adults but also in other areas; the most common sites includes thigh, shoulder, back, neck, chest, arm, and abdominal cavity (96–98). The tumor is rarely described in the dermatology literature because occurrences in superficial locations where a dermatologist would likely be consulted, such as on the scalp (99) or forehead (100), are rare. Clinically, hibernomas are indistinguishable from common lipomas. They are slowly growing and painless, typically occurring in the subcutis or rarely in muscle, and are often noted several years before excision. Although usually asymptomatic, they are occasionally tender. Overlying skin may have increased warmth (101). This benign, moderately firm, solitary, subcutaneous, tan, or red-brown tumor is generally from 5 to 10 cm in diameter, although examples up to 20 cm have been reported. The MRI finding of a lesion that is diffusely slightly hypointense to surrounding subcutaneous fat should prompt consideration of hibernoma in the differential diagnosis (102) (Fig. 34-17A). An intensely flourodeoxyglucose (FDG)-avid lesion on positron emission tomography/computed tomography (CT) examination, it can mimic a metastasis (e.g., melanoma) (103).
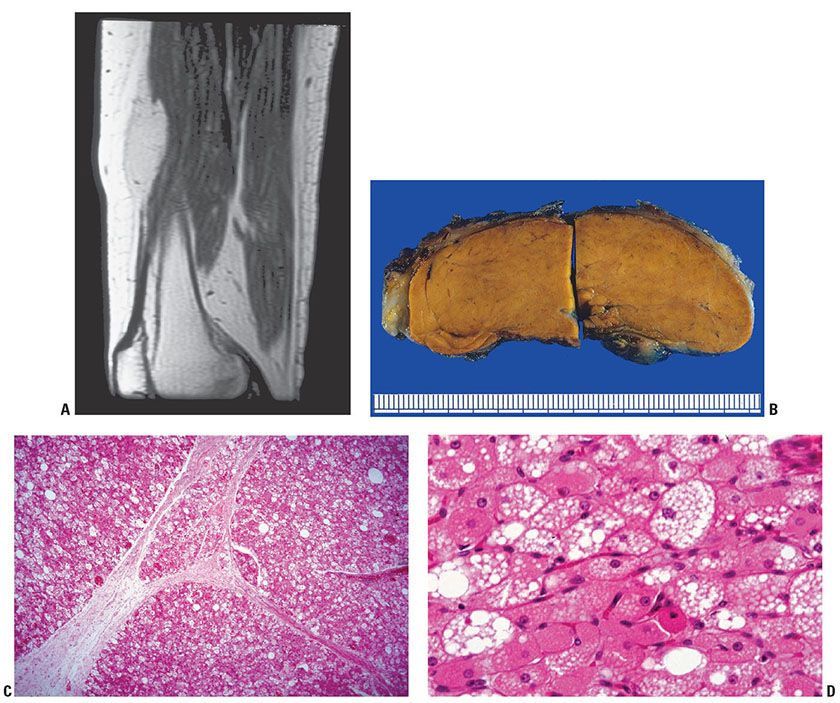
Figure 34-17 Hibernoma (69-year-old woman; 2- to 3-year history of slowly growing thigh mass). A: A longitudinal MRI of the thigh discloses a fusiform tumor between subcutaneous fat and muscle with less T1 signal than mature fat. B: The characteristic brown color of this hibernoma contrasts with the pale yellow color of attached pericapsular white fat on each end of it. It results from the prominent vascularity and many mitochondria in tumor cells. C: A low-magnification view displays the distinct lobular pattern characteristic of hibernoma. D: At higher magnification, hibernoma cells show varying degrees of complete modulation to signet-ring fat cells. These cells range from uniform round to oval granular eosinophilic cells to multivacuolated cells with multiple small Oil Red O–positive lipid droplets and centrally placed nuclei to intermixed univacuolar cells with one or more large lipid droplets and peripherally placed nuclei (lipocytes).
On gross cross section, the well-circumscribed, somewhat lobulated mass varies from tan to a deep red-brown. The distinct brown color of the gross cut surface (Fig. 34-17B) is due to prominent vascularity and a profusion of mitochondria in the granular tumor cells. Like lipomas, hibernomas have little tendency to recur locally after excision, although continued enlargement has been reported after 70% removal of an 11-cm infraclavicular tumor (104). A malignant counterpart of hibernoma (105) remains speculative.
Histopathology. This tumor is divided into numerous lobules by well-vascularized connective tissue (Fig. 34-17C). In contrast to ordinary lipoma of white fat, the highly vascular nature of the tumor is readily apparent in most sections. The tumor cells are round or polygonal and are closely apposed to one another within the lobules (Fig. 34-17D). Three principal types of cells in varying proportions can be recognized (106): (a) a small cell (average, 37 μ) with centrally placed nuclei, granular, eosinophilic cytoplasm and with or without small Oil Red O–positive lipid droplets and distinct cellular membranes; (b) a larger, multivacuolated fat cell (average, 54 μ) with scanty granular, eosinophilic cytoplasm, referred to as a mulberry cell; and (c) a still larger, univacuolated fat cell (average, 64 μ) with peripherally placed nucleus, variably present and perhaps predominating, making distinction from ordinary lipoma difficult. The nuclei of the granular and multivacuolated cells are usually centrally located, and the nuclei of the univacuolated fat cells are peripheral. Cells of the three principal types, with transitional forms, usually are dispersed randomly throughout the lobules. In most tumors, multivacuolated mulberry cells predominate. Some tumors, however, contain a few lobules, particularly at their peripheries, that are composed entirely of univacuolated cells (106). The vacuoles in both the multivacuolated and univacuolated cells stain positively with Sudan Black or Oil Red O. Nuclear pleomorphism or hyperchromatism and lobate nuclei are rarely seen (105) and mitoses are absent. On the basis of the nature of the stroma and the appearance of the multivacuolated cells, four categories of hibernomas are described: typical, lipoma-like, spindle cell, and the rare myxoid variant (107). Ultrastructurally, the tumor cell is invested by a basal lamina. There is an inverse relationship between lipid droplet size and the number of mitochondria per unit of cytoplasm that conspicuously lacks membrane systems. Its pleomorphic mitochondria have dense matrices and transverse lamellar cristae, micropinocytotic vesicles, and periodic short plasmalemmal densities (106).
Pathogenesis. The term hibernoma reflects the fact that these tumors are composed of cells that resemble the brown fat of hibernating animals. The incomplete maturation of the mulberry cells may be attributable to underdevelopment of enzyme systems. Characteristic cytogenetic abnormalities of 11q13-21 (108) and 10q22 (109) are found in hibernomas.
Differential Diagnosis. Rhabdomyoma and granular cell tumor should be thought of before diagnosing hibernoma. Immunohistochemically, rhabdomyomas are strongly positive for myogenic markers (desmin, actin, and myoglobin) but negative for S100, while hibernoma and granular cell tumor strongly express S100. Hibernomas without lipid vacuoles are to be distinguished from granular cell tumors. Hibernomas with vacuoles should be distinguished from myxoid and round cell liposarcoma, which usually contain diagnostic multivacuolar lipoblasts. The larger, univacuolated fat cell may predominate, making distinction from ordinary lipoma difficult.
Principles of Management. Adequate treatment consists of complete excision (99,110) sparing vital structures.
LIPOFIBROMATOSIS
Lipofibromatosis is a rare pediatric neoplasm that has been variously interpreted as a type of infantile or juvenile fibromatosis, a variant of fibrous hamartoma of infancy, and a fibrosing lipoblastoma. Although it is likely that this tumor comprises part of the spectrum of what has been referred to in the literature as infantile/juvenile fibromatosis, its clinicopathologic features and, in particular, its distinctive tendency to contain fat as an integral component, warrant separate classification as lipofibromatosis.
Clinical Summary. Involvement is more common in males with a ratio of 2:1. The patients present with a soft tissue mass from 1 to 7 cm usually involving the distal extremity such as hand, arm, leg, foot, but also less commonly found involving the trunk, or head that may be evident at birth. Radiology reveals nonenhancing bands of low signal intensity on long repetition time MRI, extending along fascial planes and with an adipose component (111). Follow-up data for 25 individuals (median follow-up period, 6 years and 7 months) found regrowth of the tumor or persistent disease documented in 17 (72%), correlating with congenital onset, male sex, hand and foot location, incomplete excision, and mitotic activity in the fibroblastic element (112).
Histopathology. Adipose tissue has a spindled fibroblastic element with focal fascicular growth, typically with limited mitotic activity. The fascicles chiefly involve the septa of fat and skeletal muscle without extensive architectural effacement of adipose lobules, as is common with conventional fibromatosis, but the tumors do entrap vessels, nerves, skin adnexa, and skeletal muscle. The fibroblastic element exhibits cytologic atypia and occasional mitoses. Small collections of univacuolated cells are often at the interface between some of the fibroblastic fascicles and the mature adipocytes.
Focal immunoreactivity is present in some tumors for CD99, CD34, alpha-smooth muscle actin, and BCL-2 and, less frequently, S100 protein, muscle actin (HUC 1-1), and EMA. No reactivity is detected for desmin (D33 and D-ER-1 clones), keratins, or CD57.
Pathogenesis. A defining cytogenetic abnormality has not been defined, but a case report of a three way t(4;9;6) translocation has been reported (113).
Differential Diagnosis. The primitive nodular fibromyxoid component of fibrous hamartoma of infancy is absent.
Principles of Management. Treatment is usually conservative as local recurrence is common due to the infiltrative growth pattern, but surgical resection is possible (111).
BENIGN LIPOBLASTOMA
Clinical Summary. Lipoblastoma and lipoblastomatosis (114) are rare benign neoplasms of fetal white fat that occur almost exclusively in infants and children (115) and typically as a stable or slowly to rapidly enlarging, asymptomatic soft lobular mass in the superficial or deep layers of soft tissue, usually on the trunk or lower extremities. Other sites of occurrence include the head, neck, retroperitoneum, trunk, back, extremities, heel (116), buttock, inguinal canal, scrotum, retroperitoneum, peritoneal cavity, and lung (117). Rarely, multifocal lipoblastoma of the face has been described (118). In a child younger than 3 months of age, images showing a predominantly fatty but heterogeneous soft tissue mass are suggestive of lipoblastoma (119). Two basic forms of benign lipoblastoma occur. The variant called lipoblastomatosis is deeply situated and poorly circumscribed, extending from the subcutis to the underlying muscle (120), and comprised mostly, but not entirely, of fat on MRI (121). The more common circumscribed lipoblastoma, in contrast, is relatively superficial and encapsulated. Both forms commonly present as a painless nodule or mass, affecting boys twice as frequently as girls. Sizes range from 1 to more than 20 cm in greatest dimension, sometimes causing dysfunction of other organ systems due to mass effect. Lipoblastomas occurring on the back can have intraspinal extension (122). These neoplasms have no malignant potential but have a tendency to recur despite presumed complete excision up to 25% of the time (123).
Histopathology. Peripheral immature lipoblasts with fat vacuoles of various sizes and central mature fat cells containing single, large fat vacuoles are characteristic (Fig. 34-18) whether the tumor is deeply or superficially situated. The lipoblasts typically contain single variably-sized vacuoles that are smaller than those found in mature fat cells. These vacuoles displace the nuclei against the cytoplasmic membrane. A few small lipoblasts contain two, three, or occasionally more vacuoles. Some benign lipoblastomas also show lipoblasts with finely vacuolar cytoplasm and centrally-placed nuclei resembling hibernoma cells. Nonvacuolated cells that are spindle shaped or stellate are observed in the mucinous stroma with a fine vascular network that is strikingly similar to myxoid liposarcoma and from which it may be histologically indistinguishable. Fibrous septa partition lobules in the circumscribed form.
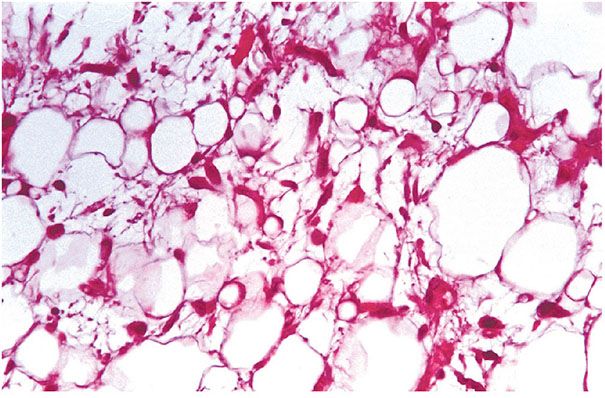
Figure 34-18 Lipoblastomatosis. The tumor is comprised of partly differentiated lipoblasts and has a prominent capillary vascular pattern with areas of mucoid matrix. Cells with large hyperchromatic nuclei are absent.
Pathogenesis. Rearrangements of chromosome 8 q11-q13 region leading to rearrangement of the PLAG1 gene are a discriminative marker that distinguishes lipoblastoma and lipoblastomatosis from myxoid liposarcoma (115). Recently, a case of lipoblastoma was found to have a t(3;8)(p13;q21.1) translocation (124). Fluorescence in situ hybridization (FISH) can serve as a decisive tool in the differential diagnosis of lipoblastoma and lipoma-like liposarcoma apart from its role in the distinction between lipoblastomas and myxoid/round cell liposarcoma (125). Confusion with typical lipoma may occur. In fact, the early occurrence of lipoblastoma, its ability to mature into a simple lipoma, its cellular composition of mainly mature adipocytes, and its benign course suggest that it may more accurately be termed an infantile lipoma (126).
Differential Diagnosis. Only the age of the patient, the abundance of lipoblasts, the absence of atypical mitoses, and a sometimes subtle lobularity distinguish benign lipoblastoma from liposarcoma, especially the myxoid type (114,127). Although liposarcomas occur very rarely in infants and young children, the distinction may be impossible by light microscopy alone. Therefore, when performing a biopsy of a childhood adipose tumor with unusual features, such as progressive or invasive growth, fresh tissue should be submitted for cell culture. The tumor karyotype will in most cases aid in differentiating lipoblastoma from myxoid liposarcoma (128).
Principles of Management. Complete resection is the only definitive treatment and should not be delayed when impingement on surrounding structures is imminent. Patients with diffuse lipoblastoma (lipoblastomatosis) are likely to have recurrent disease, usually within 2 years, and should undergo close follow-up (129) for a minimum of 5 years. Cellular maturation has been reported in serial biopsies over time. A “wait and see” approach may be justified, at least in infants with huge invasive lesions requiring a mutilating excision, since complete spontaneous resolution of a diffuse lipoblastoma in the thigh has been shown in a 2-day-old male at 1 year by MRI (130).
LIPOSARCOMA
Clinical Summary. Liposarcomas, constituting 15% to 20% of all soft tissue sarcomas, are generally deep-seated tumors of the retroperitoneum or the deep soft tissues of the extremities. Most commonly, they originate in the intermuscular fascial planes, with a special predilection for the thigh (131–133). From a fascial plane, they can extend to subcutaneous tissue. Liposarcomas are slightly more prevalent in males and the average age of onset is 50 years. Occasionally, liposarcomas arise in youth from 5 to 22 years of age, and are usually of myxoid type (134–136). Infrequently, they may be multicentric. As with most other dermal sarcomas, local recurrence is a more frequent problem than distant metastases (137). Whereas it is accepted that liposarcoma rarely occurs in the subcutaneous soft tissue, the dermis represents an exceedingly rare site of occurrence (138). Exceptionally, liposarcoma occurs primarily in the skin, presenting clinically as a dome-shaped or polypoid lesion that despite an apparent tendency to show high-grade morphologic features seems to exhibit relatively indolent clinical behavior in this location (139).
Liposarcoma is classified morphologically into three main groups: (a) well-differentiated liposarcoma (including adipocytic or “lipoma-like,” sclerosing, inflammatory, spindle cell, and their “dedifferentiated variants”); (b) myxoid and round cell (poorly differentiated myxoid) liposarcoma; and (c) pleomorphic liposarcoma. Some tumors display more than one morphologic type.
Well-differentiated liposarcoma is the type most commonly biopsied by the dermatologist. The clinical outcome of well-differentiated liposarcoma is best predicted by anatomic location (140) and is generally better than the round cell and pleomorphic types. In the subcutis, they are usually cured by local excision, rarely regrow, and do not metastasize. Therefore, the terms atypical lipoma and then later atypical lipomatous neoplasm were introduced for the subcutaneous form of well-differentiated liposarcoma (141,142). Great debate has been generated by the introduction of that term, with most now agreeing that well-differentiated liposarcoma and atypical lipomatous neoplasm should be considered synonyms describing lesions identical both morphologically and karyotypically; the term atypical lipomatous neoplasm (or atypical lipoma) should be restricted to subcutaneous tumors, especially those of small size with minimal atypical changes, and the term well-differentiated liposarcoma applied to the same histology at other deeper sites (137).
The emergence of a high-grade sarcoma pattern within a well-differentiated liposarcoma or in recurrence is known by the biologically imprecise term dedifferentiation. The clinical outcome of “dedifferentiated” liposarcoma is less aggressive than in other high-grade pleomorphic sarcomas, although it behaves worse than the usual low-grade liposarcoma with 41% of patients experiencing local recurrence, 17% with metastases and 28% ultimately succumbing to disease (138,143).
Myxoid liposarcoma and round cell liposarcoma are particularly rare in the skin (144,145), and when they do, they tend to have a more favorable outcome than their deep-seated counterparts. Myxoid liposarcoma is the most common type of liposarcoma. Clinically, myxoid and round cell liposarcoma tend to occur in the limbs, with a peak incidence ranging between the third and the fifth decade, accounting for about 30% to 35% of all liposarcomas and share both clinical and morphologic features. Metastases are common, where increased numbers of mitoses, either in the primary tumor or a local recurrence, predict metastases. Although they do metastasize to the lungs or mediastinum, extrapulmonary spread is common in myxoid liposarcoma, such as to serosal surfaces, intra-abdominal sites, skin and bone (146).
Pleomorphic liposarcoma is a high-grade pleomorphic sarcoma, containing multivacuolated lipoblasts, which usually develops during late adult life and has a propensity to metastasize. Pleomorphic liposarcoma usually occurs in the deep soft tissues and uncommonly arises in the dermis or subcutis. Despite high-grade morphology, an intradermal or subcutis tumor shows a relatively favorable prognosis with 4 local recurrences in 24 cases and no metastases or death from disease in a recent study (145).
Histopathology. Liposarcoma is diagnosed only when there is convincing evidence of synthesis and storage of fat by the tumor cells (147), the hallmark of which is the lipoblast. For a cell to be designated as a lipoblast, it must show the ability to synthesize and accumulate non–membrane-bound lipid in the cytoplasmic matrix. Acceptable malignant lipoblasts are quite variable, because they may recapitulate any stage in the normal maturation sequence. Their common morphologic denominator is a well-demarcated cytoplasmic lipid that displaces or indents one or more irregular hyperchromatic nuclei. The nucleus conforms to the contours of the lipid droplet, creating an apparent delicate scalloping of the nuclear membrane (105). Hyperchromaticity and variability from lipoblast to lipoblast support a malignant diagnosis. Despite these criteria, the distinction between malignant lipoblast and “atypical lipocyte” is at times subjective, especially in the more well-differentiated lipoma-like variants of liposarcoma. A “true” lipoblast is defined not only by a multivacuolated or univacuolated fat cell with indented or scalloped nucleus but also by its histologic context (94).
Well-differentiated liposarcoma (atypical lipoma) histologically features distended univacuolated fat cells that may vary slightly in size and shape, with occasional interspersed lipoblasts. The nuclei of the fat cells are slightly pleomorphic, and some of them are enlarged and hyperchromatic. Broad fibrous septa containing cells with enlarged hyperchromatic atypical nuclei are an important diagnostic feature (Fig. 34-19). Foci of well-differentiated smooth muscle may be found but do not influence behavior (148).
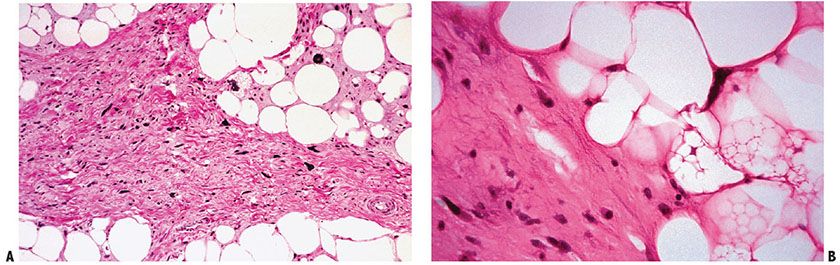
Figure 34-19 Well-differentiated lipoma-like liposarcoma (atypical lipoma). A: A broad, fibrous septum containing cells with atypical hyperchromatic nuclei partitions a tumor of fat-filled rounded cells, some of which are lipoblasts. (Courtesy of Andrew E. Rosenberg.) B: Hyperchromatic nuclei are in the fibrous septum and a dark-staining triangular lipoblast nucleus is in adjacent fat (compare with Fig. 34-4). Prolonged searching may be required to find enlarged, irregular, hyperchromatic nuclei in atypical lipoma, some indented by clear lipid vacuoles.
Transition from well-differentiated liposarcoma to nonlipogenic morphology (138) is known by the pathobiologically imprecise designation dedifferentiated liposarcoma and is limited for practical purposes to these tumors when they are located in body spaces, especially the retroperitoneum (149). The interface between the two zones is usually abrupt. A peculiar biology is displayed, which at least in most cases shows neither “anaplasia” nor “dedifferentiation” but most likely “transdifferentiation” of part of the neoplastic cells to a cellular phenotype of a different mesenchymal differentiation lineage (150). A well-differentiated (lipoma-like) liposarcoma or less commonly myxoid liposarcoma (151) will have superimposed nonlipogenic areas resembling high-grade fibrosarcoma or malignant fibrous histiocytoma with the full range of potential patterns from storiform pleomorphic and myxoid to the less common giant cell or inflammatory forms. Less common are undifferentiated large round cells resembling carcinoma or melanoma, neural-like or meningothelial-like whorls, (152,153) or myofibroblastic, rhabdomyosarcomatous, leiomyosarcomatous, even osteoblastic differentiation. “Dedifferentiated” liposarcoma rarely exhibits heterologous (most often myoid) differentiation. It is now accepted that “dedifferentiated” liposarcoma can present with low- and high-grade areas of “dedifferentiation”(143).
Spindle cell liposarcoma represents an uncommon variant of well-differentiated liposarcoma that tends to recur locally and may “dedifferentiate.” Morphologically, it is composed of a fairly bland neural-like spindle cell proliferation set in a fibrous and/or myxoid background and is associated with an atypical lipomatous component (138).
Myxoid liposarcoma (Fig. 34-20) displays various quantities of four elements: (a) proliferating lipoblasts, (b) delicate plexiform capillaries, (c) myxoid matrix, and (d) acid mucopolysaccharide lakes (Fig. 34-20A). Mitotic figures are conspicuously absent. These tumors have been subdivided into a well-differentiated type with little tendency to metastasize and a poorly differentiated type that commonly metastasizes (131). A well-differentiated myxoid liposarcoma contains, in addition to lipoblasts with spindle-shaped nuclei and several lipid vacuoles, more highly differentiated fat cells, such as so-called signet-ring cells (Fig. 34-20B), which have single large vacuoles with various amounts of lipid accumulation (Fig. 34-20C) occupying a major portion of the cytoplasm, and even mature fat cells. The tumor cells are arranged loosely in a myxoid stroma. In a poorly differentiated myxoid liposarcoma, the spindle-shaped lipoblasts have large, atypical nuclei and usually only small numbers of lipid droplets.
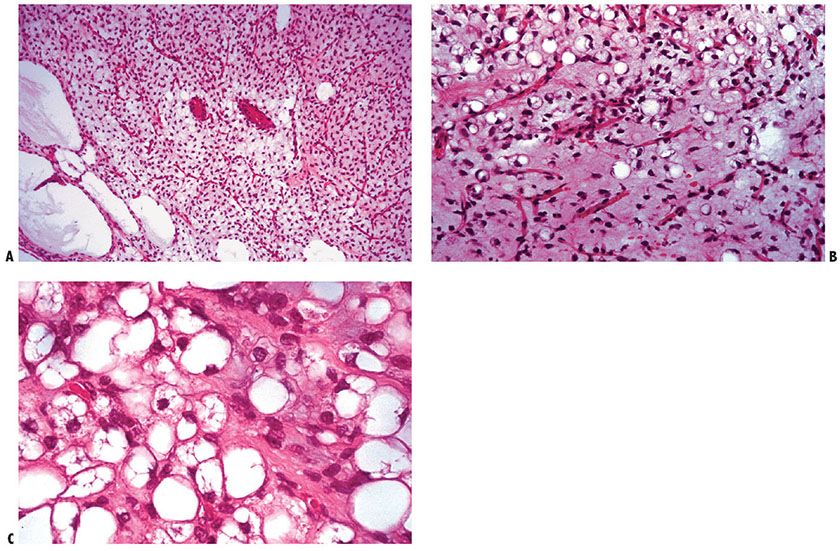
Figure 34-20 Myxoid liposarcoma. A: Angular hyperchromatic nuclei of densely packed lipoblasts might be mistaken for stellate cells of a myxoma. Pooling of myxoid material may result in a cribriform or lace-like pattern (lower left). B: Variously lipidized tumor cells in an abundant myxoid background are served by a plexiform capillary network of vessels with thinner walls than those found in a myxofibrosarcoma (myxoid “malignant fibrous histiocytoma”). C: Lipoblasts are in varying stages of cytoplasmic lipid accumulation.
Lesions combining both myxoid and round cell patterns are frequent, and wide agreement exists in considering round cell liposarcoma as the high-grade counterpart of myxoid liposarcoma (138). In general, when more than 25% of the cellular elements are round cells, the diagnosis of round cell liposarcoma should be made. However, any more than 5% round cell component in a myxoid liposarcoma portends a higher risk of metastasis or death from disease (154). As indicated by its name, round cell liposarcoma is characterized by an excessive proliferation of uniform, closely packed, rounded or oval cells, some of which contain no lipid. Others contain only small cytoplasmic vacuoles or a thin rim of clear cytoplasm (Fig. 34-21). These cells are reminiscent of Ewing sarcoma (and, similarly, may contain glycogen), malignant lymphoma, or small cell anaplastic carcinoma. Their association with other patterns of liposarcoma points to their liposarcomatous nature. Multivacuolated lipoblasts are few in number. Most tumor cells have hyperchromatic and atypical nuclei, with frequent mitoses. This tumor can be thought of as a less-differentiated modification of liposarcoma in which the cells are largely incapable of lipid accumulation. At least focal transition toward the myxoid variant, or, less frequently, a well-differentiated or pleomorphic subtype, is always present. As indicated previously, these transitions are central to the correct diagnosis of round cell liposarcoma.
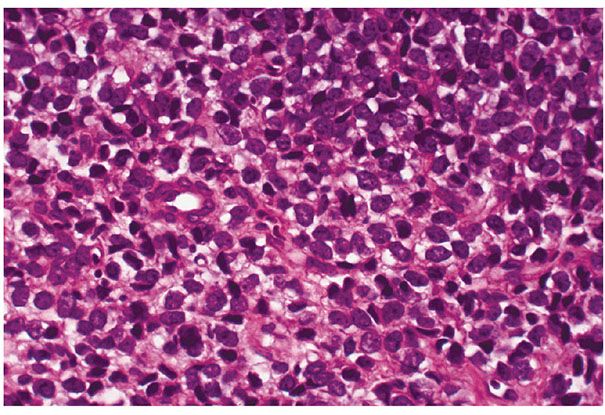
Figure 34-21 Round cell liposarcoma. Small, dark-staining tumor cells have little or no lipid accumulation. Because glycogen is involved in lipogenesis, these cells are PAS positive, and this should not be taken as evidence for Ewing sarcoma.
Pleomorphic liposarcoma is characterized by a disorderly growth pattern, an extreme degree of cellular pleomorphism, and bizarre tumor giant cells. Two histologic variants of pleomorphic liposarcoma have been described. The majority of pleomorphic liposarcomas display limited numbers of lipoblasts admixed with smaller, polygonal, round, or spindle-shaped cells with eosinophilic cytoplasm. Mitotic figures are usually rare. In any given microscopic field lacking a lipoblast, the separation of this entity from the pleomorphic storiform variant of malignant fibrous histiocytoma is difficult, if not impossible. A less common variant contains giant lipoblasts. These extremely large cells have numerous cytoplasmic lipid droplets of variable size (Fig. 34-22). Multinucleation and amphophilia of the cytoplasm causes these cells to have the appearance of atypical mulberry cells; thus, the tumor is suggestive of a “malignant” hibernoma. The histologic subgroups of pleomorphic liposarcoma have not been shown to have prognostic value, perhaps because of the limited number of cases available for study. Its karyotype is complex (155). Positivity for vimentin but not for desmin or alpha-smooth muscle actin or other myogenic markers are expected (20).
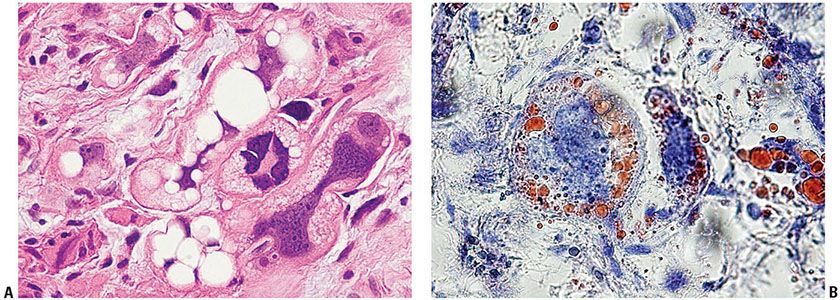
Figure 34-22 Pleomorphic liposarcoma in a 47-year-old male with 2-month history of a rapidly growing proximal arm mass medial to biceps that was superficial, subcutaneous and contacting fascia. A: Multivacuolar lipoblasts have large, irregular hyperchromatic nuclei indented by clear fat vacuoles. B: The cytoplasmic lipid content of lipoblasts, lost in routine processing, can be demonstrated by frozen sections of fresh or fixed tissue stained with Oil Red O.
Pathogenesis. Liposarcomas arise as such and for practical purposes do not develop from lipomas. DNA flow analysis shows benign and low-grade liposarcomas to be diploid and high-grade tumors to be generally aneuploid, regardless of histogenetic type (156). Well-differentiated liposarcomas are characterized by ring chromosomes derived from the long arm of chromosome 12, which results in amplification of many genes including MDM2 and CDK4; myxoid and round cell liposarcomas are characterized by a reciprocal translocation t(12;16)(q13;p11) (157) that results in fusion of the CHOP gene with the TLS gene; and pleomorphic liposarcoma is characterized by complex karyotypes (138) with a low incidence of MDM2 gene amplifications (145). A high CDK4 amplification level is a poor prognostic factor for well-differentiated and “dedifferentiated liposarcoma”(158).
Differential Diagnosis for Well-Differentiated Liposarcoma. Many other mesenchymal tumors and some carcinomas (e.g., of the kidney and adrenal) routinely contain fat, as do tumors injured by ischemia or radiation. Rarely, lymphomas can also have signet ring–like changes. Lesions such as pleomorphic lipomas and lipoblastomas illustrate that the presence of unequivocal lipoblasts do not always signal malignancy. In addition, benign fat necrosis, myxoid changes in structural fat and lipogranulomas may be in the differential diagnosis of liposarcoma. In addition, late granulomatous reactions from silicone injection can cause an incorrect diagnosis of liposarcoma (159).
Differential Diagnosis for Myxoid Liposarcoma. The differential diagnosis of myxoid liposarcoma includes benign lesions, such as myxoid spindle cell lipoma, intramuscular myxoma, and lipoblastoma, and malignant ones, such as low-grade myxofibrosarcoma (aka myxoid malignant fibrous histiocytoma) and extraskeletal myxoid chondrosarcoma. In addition, depending on the amount of myxoid stroma, there is considerable resemblance to either a myxosarcoma or a poorly differentiated fibrosarcoma, because the latter may also have a certain amount of myxoid stroma.
Differential Diagnosis for Pleomorphic Liposarcoma. Diagnostic considerations in the differential diagnosis of pleomorphic liposarcoma include pleomorphic lipoma, clear cell atypical fibroxanthoma, balloon cell melanoma, and metastatic clear cell carcinoma of renal origin.
The demonstration of neutral lipid in cells with special stains (e.g., Sudan Black, Oil Red O) on frozen sections of tumor tissue is insufficient for a diagnosis of liposarcoma, especially without the presence of lipoblasts. Historically, the main utility of immunohistochemistry in the diagnosis of liposarcoma was one of exclusion, as immunohistochemistry can be quite helpful in revealing the true identities of various sarcomas that may contain vacuolated bizarre cells, such as malignant schwannoma (S100 protein), rhabdomyosarcoma (myoglobin), and leiomyosarcoma (desmin). But smooth muscle and even rhabdomyosarcomatous differentiation (160) can be found in primary liposarcoma. Alpha-1-antitrypsin and alpha-1-antichymotrypsin can be demonstrated in “malignant fibrous histiocytoma,” regardless of subtype, although these markers may also be present in other spindle cell malignant tumors. More recently, it has been reported that the fatty acid binding protein ap2/FAB4 is a marker of adipocytes and can be used to highlight lipoblasts in poorly differentiated tumors where the diagnosis of liposarcoma is uncertain (161). In addition, strong and diffuse immunohistochemical reactions for MDM2/CDK4 are positive in well-differentiated liposarcoma and “dedifferentiated” liposarcoma (80).
By electron microscopy, cytoplasmic lipid is not membrane bound in liposarcoma, unlike in histiocytes that have ingested fat or in malignant fibrous histiocytoma (147).
Principles of Management. In aggressive (especially deep) liposarcomas, circumscription of the lesion may offer false hope of eradication by simply shelling out the tumor. In reality, liposarcomas extend microscopic pseudopod-like extensions that commonly extend along fascial planes. Biopsy and complete resection of a substantial mass of unknown type should be carefully planned, guided by MRI and radiographic findings in a manner that eliminates prior biopsy tracts and any plane potentially contaminated by surgical exposure or spreading hematoma. An additional multimodality approach, including radiation therapy and chemotherapy is often added to the therapeutic regimen (162). In addition, because of the propensity of liposarcoma, especially myxoid and round cell liposarcoma, to metastasize, thoracic, abdominal CT and bone scan are recommended in the initial evaluation as well as follow-up of high-risk patients with larger, locally recurrent or high-grade tumors of any size (146).
TUMORS OF SMOOTH MUSCLE
Smooth muscle in the skin serving as the substrate for tumors are found in arrector pili muscles, mural muscle of blood vessels, and specialized muscle in genital sites (i.e., scrotum [dartos], vulva, and nipple [areolar]).
SMOOTH MUSCLE HAMARTOMA
Clinical Summary. Smooth muscle hamartoma usually presents as a single patch that is several centimeters in diameter, on the extremities or trunk, most commonly in the lumbar region, and rarely involving the scrotum (163). It may be present at birth or may arise in childhood or early adulthood (164). Usually, there are small, follicular papules throughout the patch, although the entire lesion may be slightly elevated and/or linear (165). Transient elevation (pseudo-Darier sign) can often be elicited on rubbing (166). Associated vermiform movements are rarely seen, suggesting the lesion is acting as an innervated functional (although abnormal) unit (164). The patch shows hyperpigmentation and hypertrichosis in some patients (167) but not in others (168). If hyperpigmentation and hypertrichosis are present, an association with Becker melanosis (see Chapter 28) may exist (169). There are rare reports of multiple disseminated or generalized lesions, and familial cases, most recently reporting the presence of multiple smooth muscle hamartomas in three members of the same family (170). There is no known associated systemic involvement beyond being a potential component of the Michelin tire syndrome (see above) or malignant transformation (164). Induration, hyperpigmentation, and localized hypertrichosis diminish with time (171).
Histopathology.
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


