Topical Retinoids: Introduction
|
In topical preparations, retinoids are widely used as prescription drugs as well as cosmeceuticals. In some of these products, retinoids are naturally occurring compounds while others are synthetic molecules. All retinoids, however, share predictable pharmacology in eliciting human skin responses due to the fact that retinoid effects are primarily mediated through their intranuclear retinoid receptors acting as transcription factors. Indeed, the discovery and characterization of retinoic acid receptors (RARs) and retinoid X receptors (RXRs) have been pivotal to our understanding of the retinoid action mechanism and gave rise to the idea that retinoids act similarly to hormones.1,2 In 1976, Michael Sporn and his colleagues originally defined retinoids as both the naturally occurring compounds with vitamin A activity and the synthetic analogs of retinol. This concept is no longer adequate. Now, retinoid is defined as any molecule that, by itself or through metabolic conversion, binds to and activates the RARs, thereby eliciting transcriptional activation of retinoic acid-responsive genes that results in specific biologic responses.
Mechanism of Action
The discovery and characterization of RARs as having molecular features that are similar to steroid/thyroid hormone receptors were landmark findings.1,2 A characteristic common to these receptors is that they bind to regulatory regions in DNA called hormone response elements, or target sequences, and activate gene transcription in a ligand-dependent manner. Therefore, they are referred to as ligand (hormone)-dependent transcription factors. These receptors bind to specific DNA sequences as dimers. Unlike receptors for the steroid hormones, which form homodimers (two identical monomers); RAR, vitamin D receptor (VDR), and thyroid hormone receptors (TR) bind to their elements with greater affinity as heterodimers (two different monomers). The critical partner for heterodimerization and ultimate functioning of RAR, VDR, and TR is the RXR,3 whose physiologic ligand is 9-cis-retinoic acid.4 The ability of RXRs to interact with many receptors suggests their importance as regulatory proteins. There are three different members of RAR (α, β, and γ) and RXR (α, β, and γ), each encoded by different genes. Furthermore, each of the receptors has isoforms, adding to the diversity of retinoid receptors.
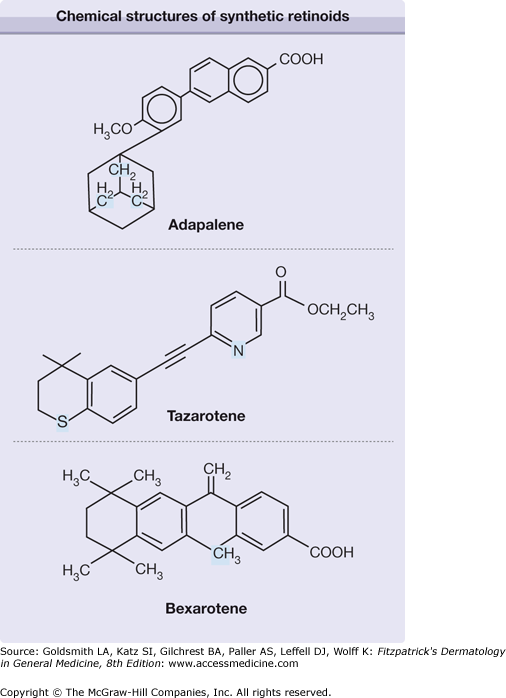
The identification and functional characterization of retinoid receptor families cleared the way for drug discovery. Many synthetic compounds have no structural similarities to all-trans-retinol or retinoic acid, yet are still considered retinoids by virtue of their ability to activate the receptor(s), therefore mediating the retinoid effect. Adapalene, tazarotene, and bexarotene are three synthetic retinoids that fit this description and have been used topically in humans (Fig. 217-1). Adapalene has restricted receptor specificity, possessing poor affinity for RAR-α, higher affinity for RAR-β and -γ, and no interaction with RXR-α.5 Tazarotene cannot directly bind to RARs or RXRs. However, its metabolite, tazarotenic acid, has receptor selectivity for RARs (RAR-β > RAR-γ > RAR-α),6 and is felt to be the principal active form. Bexarotene is an RXR-selective retinoid (rexinoid), which is 100-fold more potent at binding to RXR than to RAR receptor.7
Systematic analysis of relative levels of each RAR and RXR has been made in human skin and shows that the human epidermis expresses RAR-α, RAR-γ, RXR-α, and RXR-β messenger RNAs (mRNAs).8,9 The relative levels of retinoid receptor mRNAs mirror their relative protein levels. In nuclei from human epidermal cells, there are five times more total RXRs than RARs.9 For RAR proteins, 87% are RAR-γ and 13% are RAR-α with no detectable RAR-β. Of the RXR proteins, 90% are RXR-α. Furthermore, only RXR-α and RAR (mostly γ) heterodimers bind to retinoic acid-responsive elements (RAREs).9,10 At their physiologic levels, RARs and RXRs in human skin bind RAREs as heterodimers and transactivate these response elements in response to RAR, but not to RXR ligands. However, the RXR is key, as the heterodimer will not bind to the RARE without the presence of the RXR protein. Ultraviolet (UV) irradiation of human skin causes a rapid and significant reduction of retinoid receptors, both RAR-γ and RXR-α.11 Although this decrease in the receptor levels is transient and returns to pre-UV treatment levels within 2 days, UV irradiation results in functional impairment of receptor-dependent retinoid responsiveness in human skin. Interestingly, pretreatment of skin with all-trans-retinoic acid before UV irradiation reduces receptor loss and accelerates receptor recovery.11
Retinoid actions are mediated by retinoid receptors. Given that the receptors are transcription factors, the ultimate skin effects of retinoids (phenotypic changes) must be accomplished through regulated gene expression. The best-established mechanism by which retinoid receptors modulate gene expression is activation of retinoid target genes through direct binding to RARE in the gene promoter, thereby stimulating the basal transcriptional machinery. In skin, cellular retinoic acid binding protein (CRABP) II, cellular retinol binding protein (CRBP), retinoic acid 4-hydroxylase (CYP26), and keratin 6—all of which contain RAREs—are regulated by all-trans-retinoic acid.12–15 However, the products of these genes are unlikely to be the effectors of the retinoid response and thus cannot account for the pleiotropic effects of retinoids in skin. Undoubtedly, additional RARE-regulated genes will be identified that encode for proteins that function to modulate cutaneous growth and differentiation either directly or indirectly. A likely scenario is that these protein products, in turn, activate other non-RARE-containing genes in a cascading reaction to produce the clinical features of retinoid action in skin (Fig. 217-2).
Figure 217-2
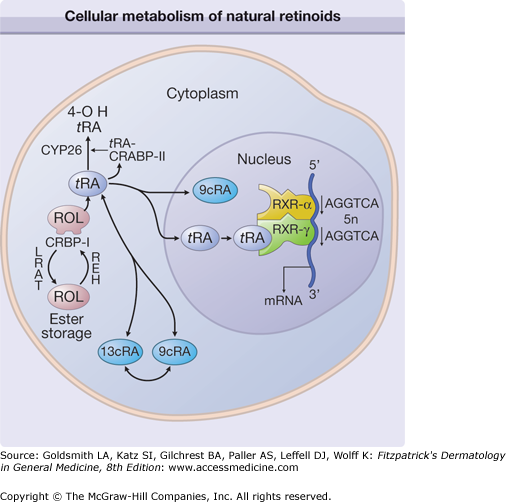
Cellular metabolism of natural retinoids and molecular mechanism of retinoid specific gene activation. Retinol (ROL) delivered to a cell is bound to cellular retinol binding protein-I (CRBP-I). ROL can be esterified, via lecithin:retinol acyltransferase (LRAT), and stored as ROL-esters. Hydrolysis of ROL-ester by its hydrolase (REH) yields free ROL. Sequential oxidation of ROL generates retinoic acid (RA), which is bound to cellular retinoic acid binding protein (CRABP). All-trans-RA (t RA) can be isomerized to 9-cis-RA (9c RA). Hydroxylation of t RA by cytochrome-P450 enzyme CYP26 generates 4-OH RA, which is relatively inactive. RARs and RXRs are intranuclear receptors for retinoids. In human skin, RAR-γ and RXR-α heterodimers bound to RA-response elements (such as AGGTCA direct repeats) transduce retinoid effects in the presence of RAR ligands. mRNA = messenger RNA.
Molecular description responsible for the most striking histologic feature of all-trans-retinoic acid-treated skin, the marked thickening of the epidermis, illustrates this point.12,16 The epidermal hyperplasia is caused by the increased proliferation of basal keratinocytes. There is also an increase in differentiation markers involucrin, loricrin, filaggrin, and epidermal transglutaminase.15 These epidermal changes collectively translate to clinical desquamation and peeling. This hyperproliferative response of epidermis to retinoic acid is mediated by its nuclear receptor.17,18 Among the genes regulated by retinoid receptors involved in the cell growth control (keratinocyte proliferation), ligands for epidermal growth factor (EGF) receptor appear to be critically important. In human skin in vivo, topical retinoic acid induces heparin-binding (HB)-EGF and amphiregulin (AR), which stimulate basal cell growth via the cell surface EGF receptor activation.19,20 Interestingly, neither HB-EGF nor AR possesses any putative RARE. Therefore, a direct modulation of HB-EGF or AR by RARE promoters seems unlikely. Rather, RAR/RXR is likely regulating other transcription factor(s) that would, in turn, regulate AR and HB-EGF gene expression (Fig. 217-3). The identity of such transcription factor(s) remains to be determined.
Figure 217-3
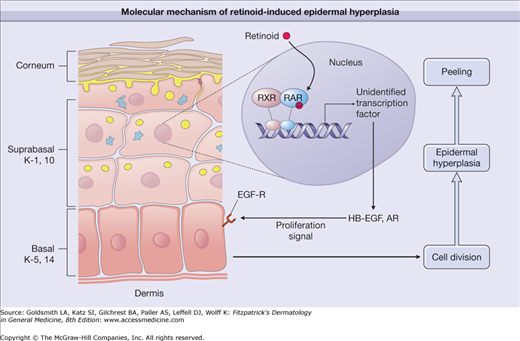
Molecular mechanism of retinoid-induced epidermal hyperplasia. Topical application of a retinoid activates retinoic acid receptor/retinoid X receptor (RAR/RXR) heterodimers in suprabasal keratinocytes, causing activation of yet unidentified transcription factors. These, in turn, activate the synthesis of heparin-binding epidermal growth factor (HB-EGF) and amphiregulin (AR). Through activation of epidermal growth factor-receptor (EGF-R), HB-EGF and AR cause proliferation of basal keratinocytes, thereby inducing thickened epidermis and consequent peeling/flaking of the stratum corneum. K = keratin.
Pharmacodynamics
All-trans-retinoic acid (tretinoin), which binds to and activates RARs, is derived from sequential oxidation of all-trans-retinol (or vitamin A) and all-trans-retinaldehyde. It is a 20-carbon molecule that consists of a cyclohexenyl ring, a side chain with four double bonds (all arranged in trans-configuration), and a carboxylic-acid end group (Fig. 217-4A). The numbering of the carbon atoms is as shown in Figure 217-4B. The terms 9-cis- (alitretinoin) and 13-cis-retinoic acid refer to stereoisomers of all-trans-retinoic acid in which the double bond that begins with the ninth and thirteenth carbon atoms, respectively, is in the cis- rather than trans-configuration. The 9-cis-retinoic acid binds to the RXR-α receptor with high affinity, being 40 times more potent than all-trans-retinoic acid on RXR-α. 9-cis-retinoic acid can also bind RAR receptors thus at times referred to as a panagonist of retinoid receptors.4,21 The fourth carbon atom is located in the cyclohexenyl ring of retinoic acid and is involved in a hydroxylation reaction to generate 4-hydroxy-retinoic acid (see Fig. 217-4C). The addition of a hydroxyl group to the cyclohexenyl ring renders the molecule more polar, making it more amenable to excretion/elimination from cells and organisms. It is also much less active biologically. 4-Hydroxy-all-trans-retinoic acid can be further oxidized to 4-oxo-retinoic acid.
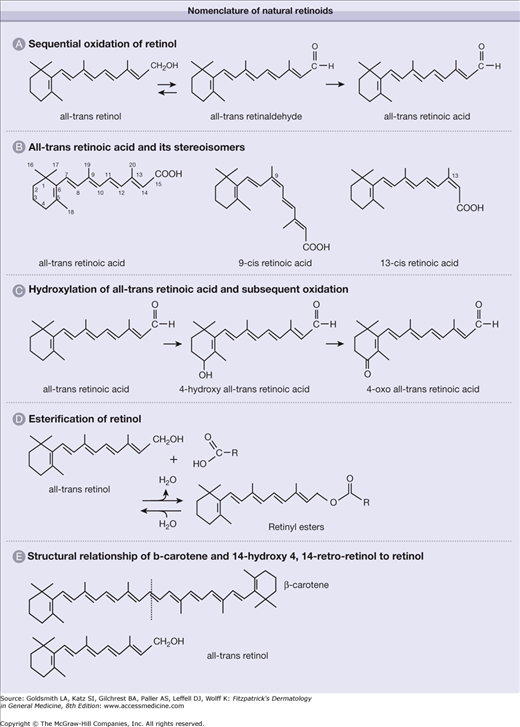
A group of compounds referred to as retinyl esters functions as the molecular storage form of retinol. The compounds are formed by esterification of retinol with fatty acids (see Fig. 217-4D), which specify the ester. For example, retinyl palmitate is generated when retinol is esterified with palmitic acid. Hydrolysis of retinyl esters regenerates retinol. Finally, β-carotene is a symmetric 40-carbon molecule (twice the number of carbons in retinol or retinoic acid) present in green and yellow–orange fruits and vegetables. As suggested by stoichiometry, one molecule of this carotenoid can potentially generate two molecules of retinol by cleavage of the central double bond (see Fig. 217-4E).
Topical treatment of human skin with all-trans-retinol increases retinyl ester levels in the epidermal layer by more than tenfold.12 This reaction is catalyzed by two enzymes: (1) lecithin/retinol acyltransferase and (2) acyl-coenzyme A/retinol acyltransferase. In human keratinocytes, lecithin/retinol acyltransferase has the predominant retinol-esterifying activity.22
Sequential oxidation of all-trans-retinol forms all-trans-retinoic acid, with all-trans-retinaldehyde as the intermediate metabolite. The first step (oxidation of all-trans-retinol to all-trans-retinaldehyde) is rate limiting for all-trans-retinoic acid formation. Topical application of all-trans-retinol to human skin results in histologic and molecular alterations that mimic those following all-trans-retinoic acid treatment.12 These include epidermal hyperplasia, epidermal spongiosis, compaction of the stratum corneum, and induction of CRBP, CRABP-II, and CYP26.22,23 In retinol-treated skin, all-trans-retinoic acid is minimally detectable and is no different from levels found in untreated normal skin. The lack of significant accumulation of all-trans-retinoic acid in retinol-treated skin is a result of the tightly regulated conversion of retinol to retinaldehyde and the effective hydroxylation of all-trans-retinoic acid that is formed by CYP26. The observation that all-trans-retinol and retinoic acid elicit similar responses in human skin, but that retinol does so without detectable increases in retinoic acid levels, indicates that endogenously synthesized retinoic acid is a much more efficient activator of retinoid pathways than exogenously supplied retinoic acid.
In human skin all-trans-retinoic acid is catabolized primarily to the more polar 4-hydroxy-all-trans-retinoic acid, which is further metabolized to 4-oxo-retinoic acid. 4-Hydroxy-retinoic acid is tenfold less potent in inducing retinoid responses in human keratinocytes and mouse skin than all-trans-retinoic acid.24 In untreated normal human skin, CYP26 activity is minimally detectable. Topical administration of all-trans-retinoic acid or all-trans-retinol to human skin, however, increases its activity several fold.23,25 This hydroxylase activity is also induced in skin treated with 9-cis- or 13-cis-retinoic acid, but remarkably, it has substrate specificity for the all-trans-retinoic acid isomer only and does not hydroxylate 9-cis- or 13-cis-retinoic acid, retinol, or retinaldehydes.23 With all-trans-retinoic acid, CYP26 activity is maximally induced after 24 hours of occlusive treatment. The CYP26 activity can be effectively inhibited by ketoconazole and a related azole, liarozole.16,25 By blocking this major inactivation pathway of all-trans-retinoic acid, topical liarozole can serve as a retinoic acid mimetic or amplify human skin responses to all-trans-retinol and all-trans-retinoic acid.16 This approach of targeting CYP26 has given rise to a novel class of new chemical entities referred to as retinoic acid metabolism blocking agents
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


