Thermoregulation: Introduction
|
The Role of Skin in Human Thermoregulation
Human beings are homeotherms: we maintain our internal, or core temperature of the body within a narrow range despite thermal stresses. Thermal stress can arise from variations in environmental temperature or from the human body itself, as with heat generation by skeletal muscle during dynamic exercise. When thermal stresses arise from the environment, changes in skin temperature occur prior to any change in internal temperature. When thermal stresses arise from the body itself as with exercise, changes in core temperature occur prior to any change in skin temperature. In either case, thermal gradients are established between the skin and the body core. If skin temperature is lower than core temperature, heat will be lost from the body unless skin blood vessels constrict. If skin temperature is greater than core temperature, the body will gain heat, unless skin vessels dilate and sweat glands produce perspiration. The skin is thus a crucial component of human thermoregulation.
Human thermoregulation is achieved through an integration of several physiological processes. These integrated processes make up thermoregulatory reflexes that maintain a stable internal temperature at a “set point” of 37°C (98.6°F) despite thermal stresses. The set point is not invariant and may fluctuate as much as 0.5°C–1.0°C (0.9°F–1.8°F) according to circadian rhythms and during the menstrual cycle in females. The thermoregulatory reflexes designed to preserve internal temperature at the set point are coordinated by thermally sensitive neurons in the anterior hypothalamic–preoptic area and spinal cord, which respond to the changes in internal and skin temperatures. For example, cold-sensitive neurons in the anterior hypothalamic–preoptic area and spinal cord integrate afferent sensory inputs and activate heat-conserving mechanisms that include cutaneous vasoconstriction and increased metabolic heat production (shivering). Conversely, the stimulation of heat-sensitive neurons in the anterior hypothalamic–preoptic area and spinal cord integrate afferent sensory inputs and activate heat-dissipating mechanisms that include cutaneous vasodilation and sweat production.
Heat Transfer
To maintain thermal balance, heat gained or lost by the body must equal heat dissipated from, or produced by the body. This concept can be mathematically expressed as:
- ΔS = change in heat storage by the body
- M = metabolic heat production and is defined as the rate of transformation of chemical energy into heat and mechanical work
- E = evaporative heat loss and is defined as the rate of heat loss by evaporation of water from skin and surfaces of the respiratory tract. E is dependant on (1) rate of sweat secretion, (2) water vapor pressure of the environment, and (3) the area of evaporative surface.
- R = radiant heat gain or loss and is defined as heat exchange by emission and absorption of electromagnetic (infrared) radiation. This component accounts for 50%–60% of the heat loss in a thermally comfortable (thermoneutral) individual, but can easily become a net heat gain as when one is in direct sunlight.
- C = convective heat gain or loss and is defined as heat exchange due to forced movement of a fluid, either liquid or gas. This component is responsible for the transfer of heat to the skin through skin blood flow and transfer from the skin to the environment by air or water movement. Within the body, the cardiovascular system is the major mediator of convective heat transfer. C is dependent on (1) body surface area, (2) temperature differences, and (3) fluid (or air) movement.
- K = conductive heat gain or loss and is defined as heat transfer by flow down a temperature gradient, as between tissues and blood, between blood and skin, and between skin and the environment. This is usually combined with convective heat transfer.
- W = useful mechanical work
The sum of R, C, and K is determined by the temperature gradient between the skin and the environment. If ΔS is zero, the body is in heat balance. This “thermoneutral” condition is characterized as having low skin blood flow of approximately 5% of cardiac output. Sweating does not occur during thermoneutrality.
If ΔS < 0, the body is losing heat, core temperature is falling, and thermoregulatory reflexes are evoked to conserve heat. Thermal stability is maintained through reduction of skin blood flow that can approach zero during maximal vasoconstriction. Reduction in skin blood flow increases the thermal insulation between the body and the environment by minimizing losses through conductive (K), convective (C), and radiant (R) mechanisms. If heat loss continues despite low skin blood flow, metabolic generation of heat (M) through the shivering of skeletal muscle is initiated to restore and maintain core temperature. Brown adipose tissue can also be a source of metabolic heat generation through nonshivering thermogenesis.1 Although originally thought to be important only in human neonates where brown adipose tissue is 2%–5% of body weight, some brown adipocytes persist into adulthood.2 These adipocytes can directly generate heat (M) to maintain core temperature.3 If, despite all these mechanisms, ΔS remains negative, core temperature will fall and life-threatening hypothermia may result.
If ΔS > 0, the body is gaining heat and core temperature is rising. Under this circumstance of heat stress, thermal stability is maintained by increases in skin blood flow to facilitate heat loss through K, C, and even R losses. If heat gain continues despite these mechanisms, sweating is evoked to increase heat loss through evaporation (E) of perspiration. If ΔS remains positive, blood flow is diverted from skeletal muscle and gastrointestinal beds, providing for dramatic increases in skin blood flow. Sweat rate will also increase until maximal levels are achieved. If, despite maximal skin blood flow and maximal stimulation of sweating, ΔS remains positive, core temperature will rise and life-threatening hyperthermia, i.e., heat stroke, will occur.
Thermoregulation and the Skin
The critical role of the skin in human thermoregulation is well understood: thermoregulation is achieved through variations in blood flow and sweat production so as to maintain thermal stability.4 Without these variations, thermal stability cannot be maintained resulting in risk of hypothermia or hyperthermia (see Chapters 94 and 95). Under normothermic conditions, skin blood flow ranges from 30–40 mL/min/100 g of skin in resting humans. However, the cutaneous vasculature is exceedingly compliant so that skin blood flow can vary from nearly zero during cold stress periods with maximal vasoconstriction to 8 L/min over the body’s surface during maximal vasodilation in heat stress.5
Blood vessels in the skin are arranged in several plexuses in superficial and deep layers parallel to the skin surface. Most vessels are in the superficial layer and consist of high-resistance terminal arterioles, papillary loops, and postcapillary venules. Papillary loops are true capillaries. Blood flow through the loops is controlled by highly innervated arterioles. The loops are located near the dermal–epidermal junction, a region characterized by a maximal thermal gradient because of its proximity to the skin surface. Since the papillary loops also have a large surface area, blood flow through these vessels is a major determinant of heat exchange through vasodilation during heat stress and vasoconstriction during cold stress.
While papillary loops are found in both glabrous (palms, plantar aspect of feet, and lips) and nonglabrous skin (most of the body’s surface, including the limbs, head, and trunk), arteriovenous anastomoses (AVAs) are found mainly in glabrous skin. They represent direct connections between arterioles and venules that bypass the high-resistance arterioles and capillaries of the papillary loops. AVAs have thick muscular walls with rich noradrenergic innervation and lie deep to papillary loops.6 Because of their deeper location in the dermis and smaller surface area, AVAs are less efficient in heat transfer than papillary loops. While AVAs dilate in response to heat stress and constrict during mild-to-moderate cold stress, their major role is to mediate local vasodilation during prolonged cold exposure. AVA vasodilation delivers warm blood to maintain tissue temperature and thus tissue viability through “cold-induced vasodilation.”7
Sweat glands also play a major role in human thermoregulation (see Chapter 83). The critical thermoregulatory role of the eccrine sweat glands that are found over most of the body surface is well known. Clearly, the main function of eccrine sweat glands is to increase heat loss through the evaporation of sweat. The density of these glands varies from 700 glands per cm2 in planar and plantar skin to 64 glands per cm2 on the back8; these glands may hypertrophy with repeated heat exposure.9 Each gland is made up of a secretory coil found in the dermis with a duct that extends through the dermis and epidermis to the surface of the skin. Sweat is secreted as an isotonic fluid by the coils. NaCl is reabsorbed within the ducts so sweat that is finally delivered to the surface is hypotonic.10 Each liter of sweat evaporated is capable of removing 580 kcal from the body. Although apocrine sweat glands have been dismissed as “atavistic scent glands,” this has recently been questioned.11 Apocrine glands are usually associated with hair follicles and are most developed on the scalp, face, upper back, and chest. It has been proposed that sebum from apocrine glands acts as a surfactant at high temperatures and, thus, facilitates dispersion of eccrine sweat over the skin’s surface. At low temperatures, sebum may function to repel water from the skin and, thus, reduce heat loss.
Cutaneous circulation is a major effector of human thermoregulation.4 During heat stress, elevated internal temperature and skin temperature lead to cutaneous vasodilation through neural mechanisms and the local effect of higher temperatures on the skin vessels themselves. During the periods of cold stress, reduced temperatures mediate a cutaneous vasoconstriction through neural as well as local vascular effects. Under normothermic conditions, skin blood flow averages approximately 5% of cardiac output; however, the absolute amount of blood in the skin can vary from nearly zero during periods of maximal vasoconstriction in severe cold stress to as much as 60% of cardiac output in severe heat stress.5
Heat dissipation through the secretion and evaporation of eccrine sweat is critical to maintaining thermal stability in hot environments or during heat stress induced by strenuous dynamic exercise. Indeed, when environmental temperature exceeds blood temperature, the evaporation of sweat is the sole mechanism for heat dispersal. Sweat secretion is controlled primarily by sympathetic cholinergic nerves that release acetylcholine (Ach) to activate muscarinic receptors on the glands. Sweat secretion can be augmented by local production of nitric oxide near sweat glands.12 Stimulated glands produce an isotonic fluid that becomes progressively hypotonic as the Na+ is reabsorbed in the sweat gland duct by active ion transfer (see Chapter 84).
In glabrous skin, cutaneous arterioles are innervated by sympathetic vasoconstrictor nerves that release norepinephrine and other cotransmitters.7,13–16 All thermoregulatory reflex changes in blood flow in these areas are caused by changes in noradrenergic vasoconstrictor activity and the effects of local temperature on the skin blood vessels themselves (Fig. 93-1).4,7,17
Figure 93-1
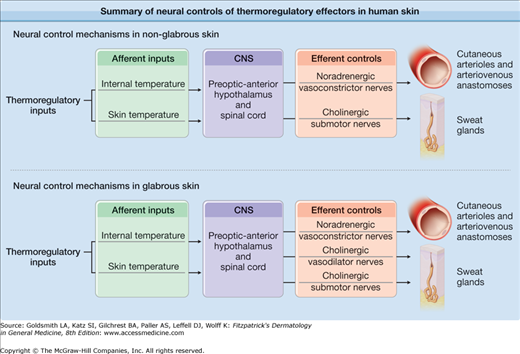
Summary of neural controls of thermoregulatory effectors in human skin: Thermoregulatory reflexes are mediated by the afferent inputs of internal and skin temperature. These afferent inputs are integrated in the preoptic anterior hypothalamus and spinal cord areas of the central nervous system. Efferent control of blood vessels and sweat glands in glabrous skin is mediated by noradrenergic vasoconstrictor nerves and cholinergic sudomotor nerves, respectively. In nonglabrous skin regions, efferent control of blood vessels is effected through a system of dual sympathetic innervation, noradrenergic active vasoconstrictor nerves and cholinergic active vasodilator nerves. Sweat glands also receive cholinergic sympathetic innervation; however, whether cholinergic active vasodilator nerves and cholinergic sudomotor nerves are one and the same is not known.
In nonglabrous skin, changes in skin blood flow are mediated by two branches of the sympathetic nervous system: (1) noradrenergic vasoconstrictor nerves as found in glabrous skin and (2) a cholinergic active vasodilator system.4,7,17 These dual sympathetic neural control mechanisms are the major effectors of thermoregulatory responses. Vessels in nonglabrous skin also respond to the effects of local temperature changes (Fig. 93-2).4,7,17
Figure 93-2

Skin blood flow responses to cold stress and heat stress in nonglabrous skin: under normothermic conditions, skin blood flow is relatively low (approximately 5% of cardiac output) and skin vessels receive relatively minor neural inputs from active vasoconstrictor and vasodilator nerves. During cold stress, reductions of skin and internal temperatures lead to reflex increases in sympathetic noradrenergic vasoconstrictor nerve activity. Increased vasoconstrictor system tone reduces skin blood flow, thus increasing thermal insulation and conserving body heat. During heat stress, increasing skin and internal temperatures lead to reflex increases in sympathetic cholinergic active vasodilator nerve and cholinergic sudomotor activity. Increased activity of the vasodilator system leads to potentially dramatic increases in skin blood flow that can reach 60% of cardiac output. High skin blood flow delivers heat to the skin surface where it is removed to the environment primarily through evaporation of sweat.
In normothermia, cutaneous arterioles are under little neural tone. During cold stress, reduction of skin temperature and/or internal temperature cause a thermoregulatory reflex-mediated reduction in skin blood flow to conserve body heat. Enhanced noradrenergic vasoconstrictor tone mediates an arteriolar vasoconstriction and, thus, decreases skin blood flow.
Conversely, during heat stress, thermoregulatory reflexes that facilitate body cooling are affected. As internal temperature continues to rise over a threshold value of approximately 37°C (98.6°F), a cutaneous vasodilation begins. At this threshold, active vasodilator tone to the cutaneous arterioles is enhanced. At rest, sweating also begins at the same internal temperature threshold. Vasodilator tone increases as internal temperature increases. Enhanced vasodilator activity decreases smooth muscle tone, leading to an arteriolar vasodilation, and, thus, an increase in skin blood flow, especially through the papillary loops. High skin blood flow delivers heat to the body surface where it is dissipated to the environment in conjunction with the evaporation of sweat. Overall, the active vasodilator system is responsible for 80%–95% of the elevation in skin blood flow that accompanies heat stress. A small, but significant portion of the vasodilation is mediated by the direct vasodilator effects of local heat on the cutaneous vessels.18
Dual vasoconstrictor nerves and vasodilator nerves in skin were first suggested in 1931 by Lewis and Pickering19 and confirmed by Grant and Holling.20 They measured skin temperature as an index of blood flow in the human forearm and found that large increases in response to heat stress could be abolished by sympathectomy or nerve blockade. They noted that while sympathectomy or nerve blockade caused only a slight cutaneous vasodilation during normothermia, heat stress elicited a much greater increase in skin blood flow. In addition, nerve blockade during established heat stress abolished any cutaneous vasodilation. These results suggested that cutaneous vessels in nonglabrous skin are innervated by sympathetic active vasodilator as well as sympathetic vasoconstrictor nerves. In the 1950s, their findings were confirmed by Edholm et al21 and by Roddie et al.22 In addition, it has been shown that bretylium tosylate (a prejunctional noradrenergic neuronal blocking agent) abolishes the cutaneous vasoconstriction induced by cold stress, but does not alter the vasodilator responses induced by heat stress.23 This confirmed that dual efferent neural systems control the cutaneous arterioles: a noradrenergic vasoconstrictor system and a nonadrenergic active vasodilator system.
Cutaneous Responses to Heat
 Despite the fact that the cutaneous active vasodilator system has been studied for many decades, the specific mechanisms by which the cutaneous active vasodilator system functions are only partly understood.19,20,24 Several hypotheses have been proposed for how active vasodilation in skin works, but none has been completely proven.
Despite the fact that the cutaneous active vasodilator system has been studied for many decades, the specific mechanisms by which the cutaneous active vasodilator system functions are only partly understood.19,20,24 Several hypotheses have been proposed for how active vasodilation in skin works, but none has been completely proven.
 In the initial descriptions of cutaneous active vasodilation, it was noted that sweating and active vasodilation began at about the same time in a resting person subjected to heat stress.20 This observation led to the hypothesis that the mechanism of cutaneous active vasodilation involved cholinergic sudomotor nerve activity.20,25–27 Additional evidence that favored a close mechanistic relationship between active vasodilation and sweating comes from observations of persons with anhidrotic ectodermal dysplasia and Ross Syndrome. Persons with this congenital anhidrotic ectodermal dysplasia lack sweat glands (see Chapter 83), do not sweat nor do they have cutaneous active vasodilator responses to heat stress.27 Persons with Ross syndrome have an acquired loss of cutaneous cholinergic neurons and, hence, do not sweat or vasodilate skin during heat stress.28 Patients with either of these rare conditions are extremely heat intolerant. Despite observations such as this, the precise relationship between sweat glands and active vasodilation remains poorly defined. While it is clear that cholinergic sudomotor nerves control sweat glands, whether sudomotor and vasodilator nerves are one and the same or separate systems is unknown.29,30
In the initial descriptions of cutaneous active vasodilation, it was noted that sweating and active vasodilation began at about the same time in a resting person subjected to heat stress.20 This observation led to the hypothesis that the mechanism of cutaneous active vasodilation involved cholinergic sudomotor nerve activity.20,25–27 Additional evidence that favored a close mechanistic relationship between active vasodilation and sweating comes from observations of persons with anhidrotic ectodermal dysplasia and Ross Syndrome. Persons with this congenital anhidrotic ectodermal dysplasia lack sweat glands (see Chapter 83), do not sweat nor do they have cutaneous active vasodilator responses to heat stress.27 Persons with Ross syndrome have an acquired loss of cutaneous cholinergic neurons and, hence, do not sweat or vasodilate skin during heat stress.28 Patients with either of these rare conditions are extremely heat intolerant. Despite observations such as this, the precise relationship between sweat glands and active vasodilation remains poorly defined. While it is clear that cholinergic sudomotor nerves control sweat glands, whether sudomotor and vasodilator nerves are one and the same or separate systems is unknown.29,30
 An alternative hypothesis with strong support proposes that cutaneous active vasodilation is mediated by a complex cotransmitter system (eFig. 93-2.1). This proposal is based on studies of an atropine-resistant, cholinergic cotransmitter system in the cat paw that relies on corelease of acetylcholine (Ach) and vasoactive intestinal peptide (VIP) to accomplish vasodilation.31 Based on these observations, it was proposed that a single set of neurons in human skin could control both active vasodilation of skin arterioles and sweating by releasing both Ach and the neuropeptide cotransmitter VIP. Ach would cause sweating and VIP would cause active vasodilation.31 This atropine-resistant cotransmitter mechanism could explain why atropine abolishes sweating, but not active vasodilation during heat stress in humans.4,32
An alternative hypothesis with strong support proposes that cutaneous active vasodilation is mediated by a complex cotransmitter system (eFig. 93-2.1). This proposal is based on studies of an atropine-resistant, cholinergic cotransmitter system in the cat paw that relies on corelease of acetylcholine (Ach) and vasoactive intestinal peptide (VIP) to accomplish vasodilation.31 Based on these observations, it was proposed that a single set of neurons in human skin could control both active vasodilation of skin arterioles and sweating by releasing both Ach and the neuropeptide cotransmitter VIP. Ach would cause sweating and VIP would cause active vasodilation.31 This atropine-resistant cotransmitter mechanism could explain why atropine abolishes sweating, but not active vasodilation during heat stress in humans.4,32
eFigure 93-2.1
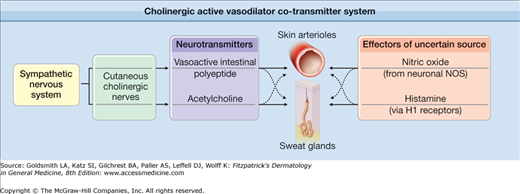
Cholinergic active vasodilator cotransmitter system: in skin, the classical neurotransmitter acetylcholine is released from cholinergic sympathetic nerves along with the neuropeptide cotransmitter vasoactive intestinal polypeptide (VIP) and perhaps other cotransmitters. These neurotransmitters cause dilation of the cutaneous vasculature and activate sweating during periods of heat stress. Nitric oxide production by the neuronal isoform of nitric oxide synthase also plays a role in vasodilation during heat stress. Activation of H1 receptors by histamine may also be involved. The precise roles and interactions of acetylcholine and the cotransmitters in mediating thermoregulatory reflex responses to heat stress remain incompletely understood.
 The cotransmission hypothesis is attractive for several reasons: (1) VIP is a vasodilator (via cyclic adenosine monophosphate [cAMP]); (2) VIP is found in human nerve endings associated with sweat glands33 and blood vessels34; and, (3) VIP is colocalized with Ach in cholinergic nerve terminals.33 VIP has also been implicated in the control of sweat glands; exogenous VIP increases muscarinic receptor affinity for methacholine (a muscarinic receptor agonist) and may thus promote sweat production as well as active vasodilation.35,36
The cotransmission hypothesis is attractive for several reasons: (1) VIP is a vasodilator (via cyclic adenosine monophosphate [cAMP]); (2) VIP is found in human nerve endings associated with sweat glands33 and blood vessels34; and, (3) VIP is colocalized with Ach in cholinergic nerve terminals.33 VIP has also been implicated in the control of sweat glands; exogenous VIP increases muscarinic receptor affinity for methacholine (a muscarinic receptor agonist) and may thus promote sweat production as well as active vasodilation.35,36
 The cotransmission hypothesis was tested in humans in a series of three studies designed to examine the involvement of cotransmission in the cutaneous active vasodilator system.37 The first of these studies confirmed the classic results of Roddie et al22 that local application of atropine to skin abolished sweating completely, but only slightly attenuated and delayed active vasodilation during heat stress. A second study showed that iontophoretic pretreatment of skin with atropine blocked all vasodilatory responses to exogenously applied Ach, demonstrating that all cutaneous vascular responses to Ach are mediated by muscarinic receptors. These two studies demonstrated that Ach could not be the only neurotransmitter that mediated active vasodilation.37 In a third study, botulinum toxin was injected into discrete areas of skin and the effect on subsequent skin blood flow responses to heat stress was evaluated. Botulinum toxin is taken up specifically by cholinergic nerve terminals and interrupts the release of all neurotransmitters from those terminals. Treatment of skin with this agent completely abolished both cutaneous active vasodilation and sweating in the treated area of skin during heat stress. This series of three studies led to the following conclusions: (1) the only functionally important cholinergic receptors on skin blood vessels are muscarinic; (2) active vasodilation and sweating are mediated by cholinergic nerves; (3) the substances causing vasodilation must include at least one neurotransmitter coreleased with Ach from cholinergic nerves.37
The cotransmission hypothesis was tested in humans in a series of three studies designed to examine the involvement of cotransmission in the cutaneous active vasodilator system.37 The first of these studies confirmed the classic results of Roddie et al22 that local application of atropine to skin abolished sweating completely, but only slightly attenuated and delayed active vasodilation during heat stress. A second study showed that iontophoretic pretreatment of skin with atropine blocked all vasodilatory responses to exogenously applied Ach, demonstrating that all cutaneous vascular responses to Ach are mediated by muscarinic receptors. These two studies demonstrated that Ach could not be the only neurotransmitter that mediated active vasodilation.37 In a third study, botulinum toxin was injected into discrete areas of skin and the effect on subsequent skin blood flow responses to heat stress was evaluated. Botulinum toxin is taken up specifically by cholinergic nerve terminals and interrupts the release of all neurotransmitters from those terminals. Treatment of skin with this agent completely abolished both cutaneous active vasodilation and sweating in the treated area of skin during heat stress. This series of three studies led to the following conclusions: (1) the only functionally important cholinergic receptors on skin blood vessels are muscarinic; (2) active vasodilation and sweating are mediated by cholinergic nerves; (3) the substances causing vasodilation must include at least one neurotransmitter coreleased with Ach from cholinergic nerves.37
 One of the current areas of interest in thermoregulatory research is the nature of the cotransmitter (or cotransmitters) that affect active vasodilation. Recent work supports the involvement of VIP as a cotransmitter in active vasodilation.38,39 This work tested whether active vasodilation is mediated, in part, by VIP coreleased from cholinergic nerves with Ach. The neuropeptide fragment, VIP10-28, was administered by microdialysis to block the effects of VIP at VPAC1 and VPAC2 receptors. This agent was chosen because it not only blocks the two major receptors for VIP, it also blocks the effects of peptide histidine methionine (PHM). These two neuropeptides share a close structural relationship, are formed from the same prepropeptide, and are both reported to be present in human skin.40 VIP10-28 attenuated (but did not abolish) the increase in skin blood flow during heat stress. The combination of VIP10-28 with atropine did not enhance the degree of attenuation achieved with VIP10-28 alone. This finding showed a role for VIP in active vasodilation; however, since the combination of muscarinic receptor with VIP receptor blockade did not differ from VIP receptor blockade only, it was postulated that additional cotransmitters may well be involved in the process.38,39
One of the current areas of interest in thermoregulatory research is the nature of the cotransmitter (or cotransmitters) that affect active vasodilation. Recent work supports the involvement of VIP as a cotransmitter in active vasodilation.38,39 This work tested whether active vasodilation is mediated, in part, by VIP coreleased from cholinergic nerves with Ach. The neuropeptide fragment, VIP10-28, was administered by microdialysis to block the effects of VIP at VPAC1 and VPAC2 receptors. This agent was chosen because it not only blocks the two major receptors for VIP, it also blocks the effects of peptide histidine methionine (PHM). These two neuropeptides share a close structural relationship, are formed from the same prepropeptide, and are both reported to be present in human skin.40 VIP10-28 attenuated (but did not abolish) the increase in skin blood flow during heat stress. The combination of VIP10-28 with atropine did not enhance the degree of attenuation achieved with VIP10-28 alone. This finding showed a role for VIP in active vasodilation; however, since the combination of muscarinic receptor with VIP receptor blockade did not differ from VIP receptor blockade only, it was postulated that additional cotransmitters may well be involved in the process.38,39
 Recent evidence suggests a role for the neurokinin Substance P in cutaneous active vasodilation in heat stress.41 Neurokinin receptors can be desensitized by repeated exposures to Substance P so that less vasodilation in response to subsequent exposures to Substance P.42 It has been shown that repeated exposure to Substance P reduced the subsequent vasodilator response to heat stress, in support of a role for neurokinin receptors and perhaps Substance P as a mediator of cutaneous active vasodilation.
Recent evidence suggests a role for the neurokinin Substance P in cutaneous active vasodilation in heat stress.41 Neurokinin receptors can be desensitized by repeated exposures to Substance P so that less vasodilation in response to subsequent exposures to Substance P.42 It has been shown that repeated exposure to Substance P reduced the subsequent vasodilator response to heat stress, in support of a role for neurokinin receptors and perhaps Substance P as a mediator of cutaneous active vasodilation.
 In the 1990s, a series of studies were done that investigated the control of skin blood flow in the rabbit ear. Rabbits (and other lagomorphs) thermoregulate by actively dilating and constricting the blood vessels in their ears. Rabbits thus provided a possible animal model for study of the dual vasomotor controls of human nonglabrous skin. The studies in the rabbit ear showed a role for nitric oxide (NO) in thermoregulatory reflex-mediated active vasodilation.43,44 This work provided the rationale to study and clarify roles for the NO system in cutaneous active vasodilation in humans.45–47 Although initial work based on intra-arterial infusions of the nitric oxide synthase (NOS) inhibitor NG-monomethyl-l–arginine (l
In the 1990s, a series of studies were done that investigated the control of skin blood flow in the rabbit ear. Rabbits (and other lagomorphs) thermoregulate by actively dilating and constricting the blood vessels in their ears. Rabbits thus provided a possible animal model for study of the dual vasomotor controls of human nonglabrous skin. The studies in the rabbit ear showed a role for nitric oxide (NO) in thermoregulatory reflex-mediated active vasodilation.43,44 This work provided the rationale to study and clarify roles for the NO system in cutaneous active vasodilation in humans.45–47 Although initial work based on intra-arterial infusions of the nitric oxide synthase (NOS) inhibitor NG-monomethyl-l–arginine (l
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


