Fig. 6.1
Human skin is structurally altered with age. Left inner arm skin from a young subject with abundant rete ridges. Right inner arm skin from an aged subject lacks rete ridges with a thinner epidermis. In addition, dermal cellularity is reduced with less dermal extracellular matrix
6.3 Intrinsic Aging
Intrinsic aging, mainly genetically programmed, presents clinically as uniform pigmentation, loss of elasticity and reduced appendage density (hair follicles, sweat and sebaceous glands; [16]). Intrinsically aged skin is characterised by fine wrinkles, a flattened DEJ and a thinning of both the dermis and epidermis. Intrinsic aging is reported to result in an overall loss of around 1 % of total collagen per adult year [17], while elastic fibres are gradually lost from the papillary dermis [18]. Reduction in total proteoglycans, which have important roles in binding water, leads to the increasingly dry skin associated with age [19, 20]. Dermal fibroblasts are reduced in number [17, 21] and functionally altered producing more matrix metalloproteinases (MMPs) and less extracellular matrix proteins [21, 22]. Mast cells are generally reported to be reduced in aged tissue [17]. However, Gunin et al. 2011 [23] report increased mast cells in aged skin which they suggest to be an important driver of tissue damage in aging. Mechanically the skin becomes stiffer and more fragile with a loss of resilience [24] making aged skin more susceptible to damage. Intriguingly, skin components are not equally affected by skin aging. Hair follicles, for example, age relatively slowly and continue to function even at advanced age [25].
6.4 Extrinsic Aging
While all tissues undergo intrinsic aging, the skin is also uniquely subject to extrinsic aging resulting from extensive exposure to environmental factors. The effects of extrinsic aging are particularly evident on the face, chest, hands and arms, which are subject to a high degree of UV exposure over a lifetime. Extrinsically aged skin is characterised by deeper wrinkles, a leathery appearance, irregular pigmentation and extensive loss of elasticity [26, 27]. At the cellular level the effects of extrinsic aging are extensive. Confusingly the epidermis of extrinsically aged skin is reported to be thickened due to hyperplasia or thinned as a result of tissue atrophy dependent on the level and duration of UV damage [28]. However, it is the changes in the dermal components of photo-aged skin which have the most profound impact on appearance. Fibrillar collagens (type I and III) are dramatically reduced and become more fragmented [29, 30], directly leading to reduced tissue tensile strength. Collagen VII anchoring fibrils are specifically lost from the DEJ along with both fibrillin 1 and fibulin 5 [31]. Severe photo-aging causes extensive profound remodelling of the elastic fibres. In young skin elastic fibres are highly ordered in structure; thick elastin-rich fibres in the deeper dermis that run parallel to the DEJ branch into perpendicular fibrillin-rich fibres in the papillary dermis. In severely photo-damaged skin, this structure is lost completely, and elastic fibre production is pathologically increased, resulting in abundant randomly orientated elastic material [31]. In contrast to intrinsically aged skin, proteoglycans are also increased and abnormally distributed as a result of extrinsic aging co-localising with the elastic fibre material [32].
6.5 Mechanisms of Aging
With the exception of severe photo damage, aged skin is characterised by a cumulative loss of components and function. Numerous theories have been proposed to drive aging (reviewed in [33]). These range from the idea that aging is due to wear and tear and an accumulation of damage with the passage of time to more specific concepts of preprogrammed life span defined by specific age-related genes. Of direct relevance to the role of hormones in the skin is the endocrine theory of aging, which places the HPA (hypothalamic pituitary axis) as a “master regulator”, signalling the termination and onset of life stages. In reality it is probably a complex accumulation of multiple different factors (i.e. aspects of each theory) that defines skin aging and in turn drives age-associated pathologies.
Aged cells are both slower to divide and compromised in their repair mechanisms. Aged fibroblasts in culture, for example display a threefold reduction in population doubling time [34], with older cells reported to have an increased G0/G1 phase proportion due to repression of cell cycle progression genes [35]. Accumulation of DNA damage is particularly detrimental to the numerous stem cell populations that reside in the skin. Accumulated damage severely impairs the function of aged stem cells ultimately leading to loss of tissue homeostasis [36]. In aged dermis, fibroblasts are reduced in both number and capacity to synthesise ECM proteins [17] contributing to age-associated atrophy. MMP overexpression is accompanied by reduced expression of the TIMP family of MMP inhibitors [37, 38].
One of the key contributors to the aging phenotype is the generation of reactive oxygen species (ROS). These highly reactive molecules cause damage to DNA, proteins and lipids via breaks, cross-links and degradation. Intriguingly, active ROS generation is emerging as an essential early signal in wound repair [39]. Mechanistically, ROS acts via AP-1 to upregulate MMP transcription and also directly activates MMPs [40–42], ultimately contributing to overall degradation of the ECM. Once activated these degradative mechanisms are maintained by a positive-feedback loop whereby MMPs are upregulated by fragmented ECM components [43].
Another central contributor to organismal aging is cellular senescence. First observed in 1961, Hayflick and Moorhead [44] reported the phenomenon of replicative growth arrest in cultured human fibroblasts. Subsequent studies revealed that the number of cell divisions at which this limit was reached is inversely proportional to the age of the donor [45], now known to be a consequence of progressive telomere shortening [46]. Telomeres form a cap of repeated DNA sequence at the ends of chromosomes; during cell division these become progressively shorter until cells can no longer divide. Senescent cells, which were originally thought quiescent, have more recently emerged as an active driver of tissue aging, secreting a number of proteases and cytokines, termed senescence-associated phenotype (SASP), which damage surrounding tissue [47]. In addition to the overexpression of proteases [37], there is also a reduction in the expression of TIMPs [38] to inhibit the actions of MMPs.
6.6 Estrogen Synthesis
All estrogens are derived from the precursor cholesterol via multiple biosynthetic steps. While the majority of circulating estrogens are gonadally derived, estrogens can also be synthesised peripherally in nonreproductive tissues such as the liver, heart, bone and skin. Specifically, the skin contains all the components required for local estrogen synthesis, including 17β-hydroxysteroid dehydrogenase (17β-HSD), 3β-hydroxysteroid dehydrogenase (3β-HSD), aromatase and 5α-reductase [48–51]. Indeed, peripheral synthesis is thought to provide the majority of estrogens post menopause, functioning as paracrine and/or intracrine factors to maintain important tissue-specific functions [52, 53]. Prior to menopause 17β-estradiol (E2) is the main gonadally derived circulating hormone. Following menopause estrone (E) plays an important role, with high levels reportedly synthesised in adipose tissue from the adrenally derived precursor DHEA [54, 55]. Estogen can also be locally regulated by interconversion between E, E2 and E3 forms or by estrogen-sulfotransferase-mediated conversion to inactive forms [56]. Thus, at any one time the local levels of active estrogens depend on finely balanced biosynthetic, metabolic and deactivation pathways. It remains unclear exactly how menopause influences peripheral estrogen synthesis. Our data from both murine models and humans suggest that estrogen deficiency leads to widespread downregulation of peripheral hormone synthetic enzymes [57, 58]. The one exception is the enzyme aromatase, which is increased in subcutaneous adipose tissue with advancing age [59].
Aromatase, a key player in peripheral hormone synthesis, is widely expressed across many tissues [60]. Its regulation is particularly complex, with the human aromatase gene containing ten validated tissue-specific promoters [61]. In the skin, as in the bone, aromatase expression is driven by the glucocorticoid responsive distal promoter I.4 [61]. It is likely that skin injury and/or disease will alter aromatase promoter usage, as is the case in breast cancer where promoter usage switches to the cAMP responsive promoter [62]. Aromatase activity can also be regulated at the level of posttranslational modification, most notably phosphorylation [63]. Skin-derived fibroblasts and adipocytes display high aromatase activity [64, 65] as do human osteoblasts [66].
6.7 Estrogen Receptors
Estrogen signals via two distinct nuclear hormone receptors, estrogen receptor alpha (ERα) and estrogen receptor beta (ERβ) encoded by separate genes (ESR1 and ESR2), located on different chromosomes. ER expression is widespread throughout the body; in some organs both receptors are expressed at similar levels, whereas in others one receptor is more highly expressed. For example, a large body of literature documents a predominance of ERα in reproductive tissues [67]. In the skin both receptors are widely expressed; however, distribution varies depending on cell type and source. Both receptors are reported in human dermal fibroblasts [68] and sebaceous glands [69]. ERβ appears to be the dominant receptor in human eccrine and apocrine glands [70] and hair follicles, being expressed in dermal papilla cells, the outer root sheath and the bulge [69]. Keratinocyte expression is more contentious. Both receptors have been reported in neonatal foreskin-derived keratinocytes with estradiol specifically upregulating ERα [71]; however, ERβ was reportedly highly expressed in human scalp skin with no ERα expression [72]. More recently still Inoue et al. [73] report significantly reduced ERβ expression in the skin of subjects over 70. By contrast, both receptors have been reported in the upper inner arm epidermis of both young and old female subjects [74]. The receptors function as homo- or heterodimers, and in this context ERβ’s function in heterodimers has been suggested to dampen ERα-mediated gene expression [75, 76]. In addition, insulinlike growth factor-1 (IGF-1) has been shown to signal through ERs in various tissues [77].
6.8 Menopause and Skin Changes
Post menopause, there is a rapid decline in the macroscopic and histological appearance of the skin with an increase in wrinkling, sagging and dryness [78, 79] (Fig. 6.2). The use of hormone replacement therapy (HRT) can prevent many of these changes, increasing extracellular matrix components, skin hydration and elasticity and reducing wrinkles [80–82]. One area that remains contentious in skin biology, as in other tissues, is the extent to which estrogen replacement can reverse detrimental changes that have already occurred and whether estrogen is beneficial in skin that is decades post menopause rather than in the perimenopause period.
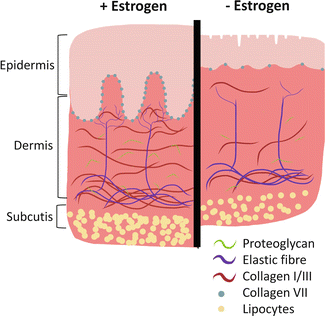
Fig. 6.2
Estrogen deficiency accelerates skin aging. Right postmenopausal estrogen deficiency leads to profound structural changes in the skin, including epidermal atrophy and wrinkling, reduced dermal elastic fibres and collagen. Collectively these changes lead to altered skin function and increased susceptibility to damage
Extensive clinical studies over recent decades have assessed the beneficial effects of HRT post menopause. The epidermis is considerably thinner post menopause [83], while HRT orally administered for 3 months or 6 months increases epidermal thickness [81, 84, 85]. Mechanistically, estrogen has been shown to directly influence keratinocyte proliferation over a far shorter period [86] and to prevent H2O2-induced apoptosis at least in vitro [87]. A large study of nearly 4,000 women found postmenopausal skin to be significantly dryer, with estrogen use functionally reversing this [78, 88]. Here HRT has been shown to significantly increase the ability of the stratum corneum to hold water [89]. More specifically, epidermal sphingolipids have been shown to be altered only in aged female skin [90], an effect that can be reversed by HRT [91, 92]. Postmenopausal changes in skin hydration are, in part, also due to changes in the dermal polysaccharides and changes in sebum levels. Here Danforth et al. [93] demonstrate that the high estrogen levels in pregnancy are linked to increased dermal hydration. Moreover, hyaluronic acid, which is known to have a high osmotic activity, is increased following estrogen treatment [94]. Sebum levels are also found to decline after menopause [95] with a decrease in both the size and activity of the sebaceous glands [96]. HRT increases the sebum level of postmenopausal skin by 35 % compared to without HRT [91].
The majority of studies have assessed the effect of menopause on dermal structure and function. The dermal extracellular matrix provides skin with many of its mechanical properties, such as resilience and tensile strength. Type I collagen is the major component of adult dermis providing tensile strength. Loss of collagen is one of the major changes implicated in the development of skin wrinkles [78, 97, 98]. Following menopause, dermal collagen content has been reported to fall by 2–5 % per year [80, 99, 100], leading to reduced dermal thickness and density. This decrease in dermal composition correlated only to postmenopausal years and not to chronological age. Affinito et al. [100] reported a decline in both collagen I and III and a specific change in the ratio of these collagen types. Brincat et al. [80] quantified the rapid loss of collagen in the initial postmenopausal period accompanied by a 1.1 % reduction in dermal thickness per year. The beneficial effects of estrogen on dermal collagen have been reported across a number of studies and trials. Topically applied estrogen increases type I and III collagen expression [101–103]. In a double-blind randomised trial, Maheux et al. [104] reported up to 30 % increase in postmenopausal skin dermal thickness following estrogen treatment. Estrogen appears to act by preferentially inducing new expression of type III collagen in aged skin, presumably invoking a “developmental”-type programme of expression [101, 105].
The skin tensile strength provided by fibrillar collagens is complemented by the elastic fibre network which endows skin with resilience, allowing it to return to a resting state following deformation. Skin resilience also negatively correlates with postmenopausal years. Using a suction device Sumino et al. [106] were able to quantify a 0.55 % decline in skin elasticity per postmenopausal year. The same study also reported a 5.2 % increase in skin forearm elasticity following 12 months of HRT. These observations are in line with previous studies that reported beneficial effects of HR on the extensibility and elasticity of forearm skin [82] and facial skin [107]. These changes in gross mechanical properties following estrogen treatment correlate with specific beneficial changes at the histological level. Punnonen et al. [108] report that topical estrogen treatment modifies the elastic fibre network, increasing elastic fibre number and improving orientation. In addition, estradiol increases cutaneous expression of tropoelastin and fibrillin proteins, key modulators of elastic fibre assembly and function [102]. By contrast, Bolognia et al. [109] report that women entering early menopause show premature degradation of elastic fibres with signs of splitting and fragmentation.
6.9 Murine Studies
Murine models provide an opportunity to mechanistically address hormonal aspects of skin aging. The ovariectomised (OVX) mouse is widely used as a model of the human menopausal state; surgical removal of the ovaries leads to a rapid decline in circulating sex steroid hormones that mimics the menopause. OVX mice are widely used to model a range of human age-associated pathologies, including osteoporosis, neurodegeneration and cardiac dysfunction, as well as perimenopausal symptoms, such as depression and hot flushes [110–112]. It is therefore surprising that comparatively little skin research has been carried out with this model. OVX is known to enhance the sensitivity of rat skin to UV-induced photo-aging, measured as wrinkling, a loss of elasticity and damage to elastic fibres [113]. This is supported by a mouse study where effects on skin extensibility and elastic recoil were measured. Here ovariectomy significantly increased recoil time in UV-exposed skin associated with elevated tissue elastase activity [114]. Very recently Fang et al. [115] have combined cryo-sectioning with AFM to reveal nanoscale changes in morphology of dermal collagen fibrils in ovine OVX samples. Other studies have explored the role of estrogen in protecting from ROS [116] and cellular senescence [117]. There are suggestions from the literature that estrogen may act as a direct antioxidant as well as induce antioxidant enzymes. Direct functional evidence was provided by Baeza et al. [118] who showed that OVX rats had increased levels of oxidised glutathione, lipid peroxidation and mitochondrial DNA damage all of which could be reversed by estrogen replacement. More recently Bottai et al. [119] report that 17β-estradiol protects both human skin fibroblasts and keratinocytes against oxidative damage.
The second major advantage of mice is their genetic tractability, crucial in the context of unravelling estrogen signalling. Mice have been generated lacking one or both of the ERs [120–125], or aromatase (ArKO), which disrupts estrogen biosynthesis [126], or more complex Cre/LoxP conditional ER nulls [121, 127, 128] and point mutants [129–131]. Phenotypic analysis of these mice had provided, and continues to provide, insight into ER-mediated physiological roles across a range of tissues (reviewed in [132]). Relevant here ER null mice display a number of skin phenotypes. First to be reported were effects on hair follicles. Moverare et al. [133] identified a role for ERα, but not ERβ, in regulating hair cycling. In a subsequent study Ohnemus et al. [134] demonstrated that ERβ does play a role, regulating catagen induction. In the same studies ERα was suggested to be important for estrogen’s effects on epidermal thickness. Markiewicz et al. [135] have recently reported increased skin collagen content in ERα null mice with decreased collagen content in ERβ nulls. Meanwhile Cho et al. [136] report a key role for ERβ in mediating UV-induced photoimmune modulation. Clearly ERs play an important role in skin homeostasis, often with directly opposite effects, supporting “ying-yang” ER interactions [75, 76].
6.10 Aging Delays Wound Healing
Under normal circumstances the skin has evolved a highly coordinated response to injury, whereby numerous cell types are sequentially activated to orchestrate rapid tissue repair (reviewed in [137]). However, with increasing age the skin not only becomes more fragile and susceptible to damage but also less able to effectively heal following injury. Indeed, numerous studies have revealed significant cellular changes in age-associated delayed healing: altered haemostasis [138, 139], reduced re-epithelialisation [140, 141], an excessive inflammatory response [142, 143] and upregulated protease activity associated with reduced matrix deposition [144]. Collectively, these age-associated changes lead to delayed acute wound healing in the elderly that predisposes to the development of chronic wounds [145]. Moreover, Wicke et al. [146] report a clinically measurable delay in chronic wound closure in those over 60 years of age versus younger patients.
6.10.1 Inflammation
The inflammatory response is delayed in elderly humans, and the overall number of inflammatory cells recruited to sites of injury is increased [142, 144, 147]. Despite these higher numbers aged macrophages are less active, with altered expression and function of TLRs [148], decreased cytokine and growth factor production [149] and reduced phagocytic activity compared to macrophages from younger counterparts [150]. Functional significance has been demonstrated in adoptive transfer experiments where macrophages from young mice are able to promote healing in aged mice [151]. Aged neutrophils are also functionally altered with reduced chemotaxis and phagocytosis [152].
6.10.2 Re-epithelialisation
Multiple studies have reported delayed re-epithelisation in aged mice and humans [140, 144, 147]. This observed delay is likely multifactorial. Key cytokines and growth factors required to stimulate keratinocyte migration and proliferation, such as PDGF, EGF and KGF, are reduced with age [153, 154]. Keratinocytes from aged donors are also intrinsically altered with reduced hypoxia-induced migration [155]. Additionally, aged keratinocytes fail to induce MMPs and other proteases required to successfully migrate through the wound granulation tissue [155]. Finally, we have recently shown that the wound-induced switch to weak keratinocyte adhesion fails in chronic wounds from aged patients [156].
6.10.3 Matrix Deposition
Fibroblasts are essential for effective repair, depositing and remodelling ECM and mediating wound contraction. Aged fibroblasts display reduced collagen synthesis [157] and impaired migration [158]. The general consensus is that aged fibroblasts are more prone to senescence with decreased proliferation with a reduced ability to respond to growth factors [159–161]. This is supported by studies indicating dysfunction in fibroblasts isolated from chronic wounds [162–164]. Intriguingly, other studies report no change in fibroblast responsiveness with age [165].
6.10.4 Angiogenesis
The literature on age-associated changes in new blood vessel formation following wounding is also conflicting. Ashcroft et al. [144] and Swift et al. [147] report increased and decreased angiogenesis, respectively, in aged mice. While this may in part reflect the different wound models used (incisional versus excisional), subsequent in vitro studies have shown that reduced angiogenic growth factors are responsible for an age-associated reduction in angiogenesis [166]. Specifically FGF2 and VEGF are decreased in the wounds of aged mice [147].
6.11 Estrogen Effects on Wound Healing
Post menopause, there is a rapid decline in wound healing ability. Studies have linked a range of menopause-associated hormones to this altered healing including progesterone [167], DHEA [168] and testosterone/DHT [169, 170]. The majority of research has, however, focussed on estrogen’s role in healing (Fig. 6.3). Studies by Ashcroft and colleagues were first to show that HRT treatment could protect against delayed healing in postmenopausal women [171]. In a subsequent study topical estrogen was shown to accelerate acute wound repair in healthy elderly men and women [172]. Shortly after came the key observation that HRT was protective against developing chronic wounds, both pressure sores (age-adjusted relative risk 0.68) and venous ulcers (age-adjusted relative risk 0.65) [173, 174]. More recently we have used microarray profiling to demonstrate that the vast majority (78 %) of genes differentially expressed in wounds from elderly versus young men were estrogen-regulated [58]. Thus, estrogen is clearly clinically and physiologically important for effective skin repair.
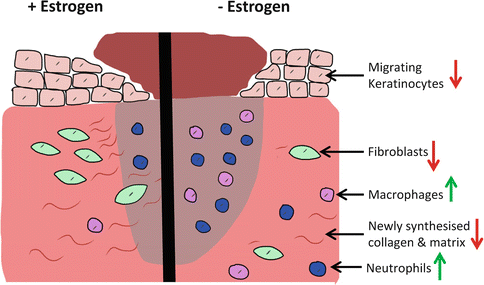
Fig. 6.3
Estrogen deficiency has a profound effect on skin wound healing. Right postmenopausal reduced estrogen leads to an inappropriately excessive but ineffective inflammatory response and retarded re-epithelialisation. Suppressed fibroblast function, decreased collagen synthesis and increased MMPs lead to a reduction in extracellular matrix
Subsequent studies focusing on estrogen’s mechanism of action have largely employed ovariectomised (OVX) rodents. OVX mice and rats display a pronounced delay in healing with altered re-epithelialisation, inflammation, ECM deposition and protease levels. Short-term estrogen replacement reverses these effects promoting keratinocyte migration, increasing matrix deposition and dampening inflammation [58, 171, 175].
6.12 Inflammation
In vitro studies have functionally linked estrogen to a range of inflammatory cells, including neutrophils, macrophages and mast, dendritic and Langerhans cells [176, 177]. In acute healing estrogen acts to dampen the inflammatory response and in human studies prevents excessive neutrophil recruitment via downregulation of L-selectin, inhibiting homing to the wound site [172]. The subsequent dampening of wound neutrophil-derived elastase levels prevents excessive degradation of ECM proteins. Estrogen also appears essential for the switch from classical (TH1; CA) to alternative (TH2: AA) macrophage polarisation. Wounds from OVX rodents contain highly CA polarised macrophages [167, 178], while estrogen treatment shifts to AA polarisation, in line with a pro-healing role [58, 167]. Estrogen is also potently anti-inflammatory in other tissues, such as the brain, where it protects against neurodegeneration [179]. Estrogen specifically dampens the expression of numerous proinflammatory cytokines, including TNF-α, MCP-1, Il-1β, Il-6 and macrophage migration inhibitory factor (MIF) [92, 180, 181]. MIF is particularly interesting and expressed by a range of wound cell types. MIF null mice are entirely resistant to the detrimental effects of OVX, implying that MIF acts as a key downstream mediator of both the detrimental effects of estrogen deficiency and the beneficial effects of estrogen replacement on skin wound healing [57, 175, 182]. Of clinical relevance, human plasma MIF levels increase post menopause and fall following HRT [57].
6.13 Re-epithelialisation
A failure of re-epithelialisation is a key aspect of both age-associated delayed acute healing and chronic wounds. Estrogen is a keratinocyte mitogen [71] directly promoting in vitro scratch wound closure in both mouse and human cells [128, 175] and in vivo re-epithelisation in murine acute wounds [183, 184]. In humans the delayed re-epithelisation associated with the postmenopausal state can be entirely reversed following 3 months of HRT [171] or short-term topical estrogen treatment [172].
6.14 Matrix Deposition and Angiogenesis
Wound collagen deposition and subsequent remodelling are delayed post menopause and promoted by HRT or topical estrogen treatment [171, 172]. Indeed in vivo estrogen promotes fibroblast proliferation and migration, with increased collagen deposition conferring increased wound strength. Estrogen directly promotes fibroblast migration demonstrated via in vitro cell assays [128, 175, 185–187] and indirectly via macrophage-produced platelet-derived growth factor (PDGF). Estrogen-driven PDGF production has a significant effect on angiogenesis, with estrogen demonstrated to be proangiogenic both in vitro and in vivo [188].
6.15 ERS and Wound Healing
Estrogen signals via two nuclear hormone receptors: ERα predominates in reproductive tissues and is strongly associated with cancer [189], while ERβ is more widely expressed in peripheral tissues [190, 191]. Estrogen’s pleiotropic role in healing is supported by widespread cellular ER expression in wound tissue and skin from a range of body sites in both mouse and human [72, 128, 192–195]. Indeed, very recently we have employed the ERE-luciferase reporter mouse [196] to directly assess ER-mediated signalling following in vivo wounding [197]. Shortly after injury ER-mediated signalling is robustly upregulated in the skin immediately adjacent to the wound. Subsequent immunohistochemical analysis revealed the signal to be predominantly localised to wound edge keratinocytes and inflammatory cells.
Crucially, in OVX mice ER-specific agonists confer entirely different effects. Treatment with the ERβ-specific agonist DPN promotes wound healing as effectively as estrogen treatment, while the ERα-specific agonist PPT has no effect. These agonist treatment effects are confirmed by estrogen replacement studies in OVX ER null mice, where ERβ null mice display a pronounced delay in healing [128]. Thus signalling through ERβ is beneficial and ERα detrimental to repair. Taking these observations further, epidermal-specific (K14-cre mediated) ER null mice were shown to phenocopy global ER nulls, and the ERβ agonist DPN was shown to directly promote keratinocyte migration in vitro, suggesting a key role for epidermal ERβ [128]. These studies are entirely consistent with clinical reports that polymorphisms in the human ERβ gene are significantly associated with venous ulceration in the Caucasian population [74, 198]. However, an alternative picture emerges for a skin flap necrosis model where the beneficial effects of 17β-estradiol on outcome are reportedly mediated via ERα [199]. The discrepancy between the two models may be explained by the observation that ERα, not ERβ, is important for controlling wound inflammation in response to IGF1 [200].
6.16 SERMs and Future Therapies
Pharmacological modulators of ERs have been developed. These include specific agonists that are experimentally invaluable and the more clinically relevant mixed agonist/antagonists, termed selective estrogen receptor modulators (SERMs). Here the SERMs tamoxifen, raloxifene and genistein have been shown to promote skin healing [183, 184]. Raloxifene acts as an antagonist in breast tissue and an agonist in bone tissue [201]. It has been shown to have a positive effect on wound healing [184]; this could be in part due to its ability to stimulate collagen synthesis in cultured fibroblasts [202] and also reduce inflammation in the wound [184]. As well as the improvement in wound healing, raloxifene is also shown to improve skin elasticity in humans following 12 months of treatment [203]. Tamoxifen was also shown to have a similar effect on wound healing reducing inflammation and restoring wound healing time in the OVX mouse to that of the intact control and estrogen treated [184]. Tamoxifen has also been studied in the formation of keloid scars in burn patients where it appears to improve scarring, decreasing collagen synthesis via reduction of TGF-β signalling and reducing fibroblast proliferation [204]. In the context of skin aging, the ERβ-selective ligand, WAY-200070, has been suggested to dampen inflammation and MMP expression in UV-damaged skin [205]. The phytoestrogen genistein has also been reported to improve gross skin changes in OVX rats [206]. In summary SERMs, which are used clinically in tissues such as bone, clearly influence physiology of postmenopausal skin; however, the mechanisms remain to be fully elucidated.
6.17 Summary
The skin undergoes widespread functional deterioration with age and following menopause. It is now widely accepted that the age-associated reduction in circulating hormones, particularly estrogen, directly accelerates skin aging, a major clinical consequence being delayed repair leading to an increased incidence of chronic wounds. Numerous studies have shown that estrogen replacement (in the form of HRT) can protect against, and possibly even reverse, skin aging, promoting repair. While macroscopic and ultrastructural aspects of these hormone-mediated effects on skin have been documented, much remains unknown. Recent studies revealing differential roles for the two estrogen receptors in skin biology and pathology have opened exciting new avenues for therapeutic intervention.
References
1.
Chuong CM, Nickoloff BJ, Elias PM, Goldsmith LA, Macher E, Maderson PA, Sundberg JP, et al. What is the ‘true’ function of skin? Exp Dermatol. 2002;11(2):159–87.PubMed
3.
Lai-Cheong JM, McGrath JA. Structure and function of skin, hair and nails. Medicine. 2009;37(5):223–6.
4.
Cotsarelis G. Epithelial stem cells: a folliculocentric view. J Invest Dermatol. 2006;126(7):1459–68.PubMed
5.
Eckert RL, Rorke EA. Molecular biology of keratinocyte differentiation. Environ Health Perspect. 1989;80:109–16.PubMedCentralPubMed
6.
Elias PM, Feingold KR. Lipids and the epidermal water barrier: metabolism, regulation, and pathophysiology. Semin Dermatol. 1992;11(2):176–82.PubMed
7.
Winsor T, Burch GE. Differential roles of layers of human epigastric skin on diffusion rate of water. Arch Intern Med. 1944;74:428–36.
8.
Maricich SM, Wellnitz SA, Nelson AM, Lesniak DR, Gerling GJ, Lumpkin EA, et al. Merkel cells are essential for light-touch responses. Science. 2009;324(5934):1580–2.PubMedCentralPubMed
9.
Holikova Z, Hercogová J, Pizák J, Smetana Jr K. Dendritic cells and their role in skin-induced immune responses. J Eur Acad Dermatol Venereol. 2001;15(2):116–20.PubMed
10.
Steplewski A, Kasinskas A, Fertala A. Remodeling of the dermal-epidermal junction in bilayered skin constructs after silencing the expression of the p.R2622Q and p.G2623C collagen VII mutants. Connect Tissue Res. 2012;53(5):379–89.PubMed
11.
Burgeson RE, Christiano AM. The dermal-epidermal junction. Curr Opin Cell Biol. 1997;9(5):651–8.PubMed
12.
Makrantonaki E, Zouboulis CC. The skin as a mirror of the aging process in the human organism – state of the art and results of the aging research in the German National Genome Research Network 2 (NGFN-2). Exp Gerontol. 2007;42(9):879–86.PubMed
13.
Kielty CM, Sherratt MJ, Shuttleworth CA. Elastic fibers. J Cell Sci. 2002;115(Pt 14):2817–28.PubMed
14.
Braverman IM, Fonferko E. Studies in cutaneous aging: I. The elastic fiber network. J Invest Dermatol. 1982;78(5):434–43.PubMed
15.
Frantz C, Stewart KM, Weaver VM. The extracellular matrix at a glance. J Cell Sci. 2010;123(Pt 24):4195–200.PubMedCentralPubMed
16.
Kohl E, Steinbauer J, Landthaler M, Szeimies RM. Skin aging. J Eur Acad Dermatol Venereol. 2011;25(8):873–84.PubMed
17.
Fenske NA, Lober CW. Structural and functional changes of normal aging skin. J Am Acad Dermatol. 1986;15(4 Pt 1):571–85.PubMed
18.
Francis C, Robert L. Elastin and elastic fibers in normal and pathologic skin. Int J Dermatol. 1984;23:166–79.
Related posts:
 Effects of Hormone Replacement Therapy on Skin Viscoelasticity During Climacteric Aging
Atrophic Vaginitis in the Menopause
Dermatologic Conditions of the Vulva During Menopause
Physical Activity and Quality of Life During Menopausal Transition and Postmenopause
Effects of Hormone Replacement Therapy on Skin Viscoelasticity During Climacteric Aging
Atrophic Vaginitis in the Menopause
Dermatologic Conditions of the Vulva During Menopause
Physical Activity and Quality of Life During Menopausal Transition and Postmenopause
 Changes to Skin with Aging and the Effects of Menopause and Incontinence
Changes to Skin with Aging and the Effects of Menopause and Incontinence
 Skin and Menopause
Skin and Menopause
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


