Introduction
Facial palsy may cause functional, aesthetic, psychological, and social consequences. The commonest cause, Bell’s palsy, will affect approximately 1 in 60 of the population over a lifetime, with almost 70% of these patients not fully recovering. Regardless of the cause, for those who do not completely recover from facial palsy, the motor sequelae has three main categories: flaccid paralysis, facial paresis, and facial synkinesis. Both flaccid paralysis and paresis may result in increased muscle activation on the unaffected side, called contralateral hyperkinesis. Botulinum toxin is frequently used to reduce the extent of visible asymmetry by reducing the activation of muscles on the healthy side. Its use on the healthy side is similar to cosmetic botulinum toxin use which principally aims to reduce the action of the target muscle. Synkinesis is the involuntary concurrent contraction of facial muscles that are usually passive during the voluntary activation of a particular muscle. Management of synkinesis is difficult due to the complex nature of muscular interactions.
Numerous theories have been put forward to explain synkinesis, including ephaptic transmission, hyperexcitability of the facial nerve nucleus, and aberrant facial nerve regeneration, but it is often a combination of many factors. Other factors such as facial co-contractions and hyperkinesis, although not a direct cause of synkinesis, contribute to its severity. In order to understand the treatment of synkinesis, it is important to first understand each of these causative and contributory factors as well as normal skeletal muscle physiology. This is explained in the next section.
Synkinesis: Pathophysiology
When the continuity of the facial nerve is disrupted due to physical (axonotmesis/neurotmesis) or physiological (neuropraxia) factors, a new axonal growth cone starts sprouting and this sometimes leads to aberrant facial nerve regeneration causing axons meant for orbicularis oris activation to grow towards the frontalis instead and causing, in this case, paradoxical frontalis activation.
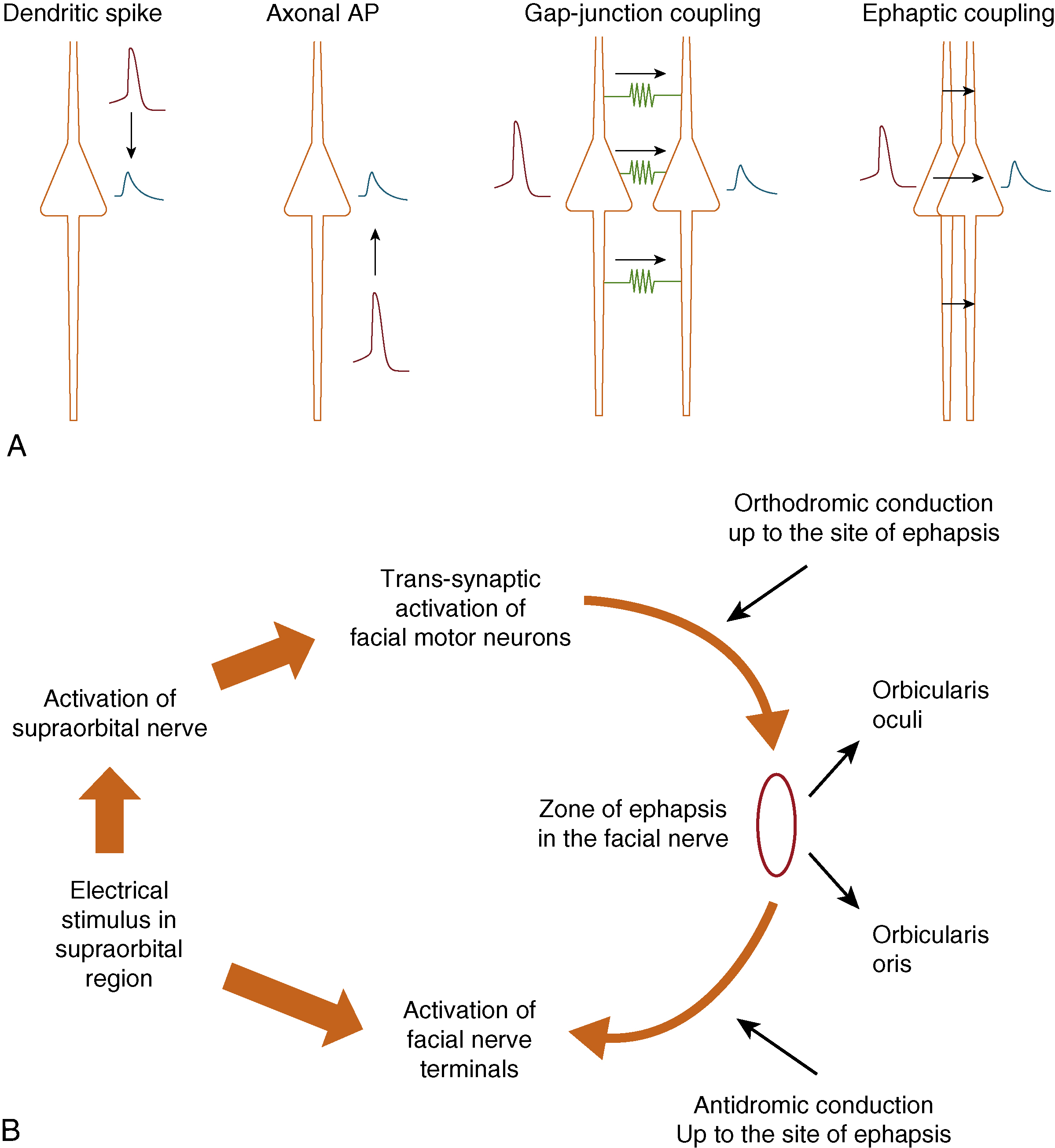
As per McKinnon et al., damage to nerves is usually mixed in that there is a combination of neuropraxia, axonotmesis, and neurotmesis. In the case of the facial nerve, this is most commonly seen in Bell’s palsy within the temporal bone. Intraneural inflammation and the subsequent swelling here causes ischemia of the nerves and subsequent scarring within certain fascicles and not within others. This “cross-wiring” then causes both orthodromic and antidromic nerve conduction to occur, thereby contributing to synkinesis as shown in Fig. 6.1 . At the same time, this conduction block forces the facial nucleus to increase its axonal firing rate, causing it to become hyperexcitable.
Simultaneously, the facial muscles (now de-innervated), undergo a gradual process of atrophy and sarcomere shortening. The latter is due to increased actin-myosin cross-linking, noted to start developing within the skeletal muscle. If the denervation period extends over 4 to 6 months, the degree of cross-linkage increases and causes the facial muscles to develop thixotrophy, a phenomenon wherein the physical properties of muscles begin to behave like viscous materials, e.g., gels. Thixotropic substances typically become less stiff when they are stretched and, in the case of hypotonic facial muscles in the initial phases of recovery, this increasing muscle stiffness (thixotrophy) can be combated by regular stretches and gentle isotonic exercises, as guided by a facial therapist. During this period, it is essential to avoid excessive muscle movements as these can lead to “eccentric” contractions as opposed to the more physiological, “concentric” contractions.
Unfortunately, due to a combination of a lack of understanding of facial therapy principles in the past as well as delayed clinical presentation, patients (typically with Bell’s palsy), have continued with active facial movements and a lack of passive lengthening, leading to the development of increased muscular stiffness. This gradually leads to sarcolemmal membrane damage, local contracture, and scarring ( Fig. 6.2 ).
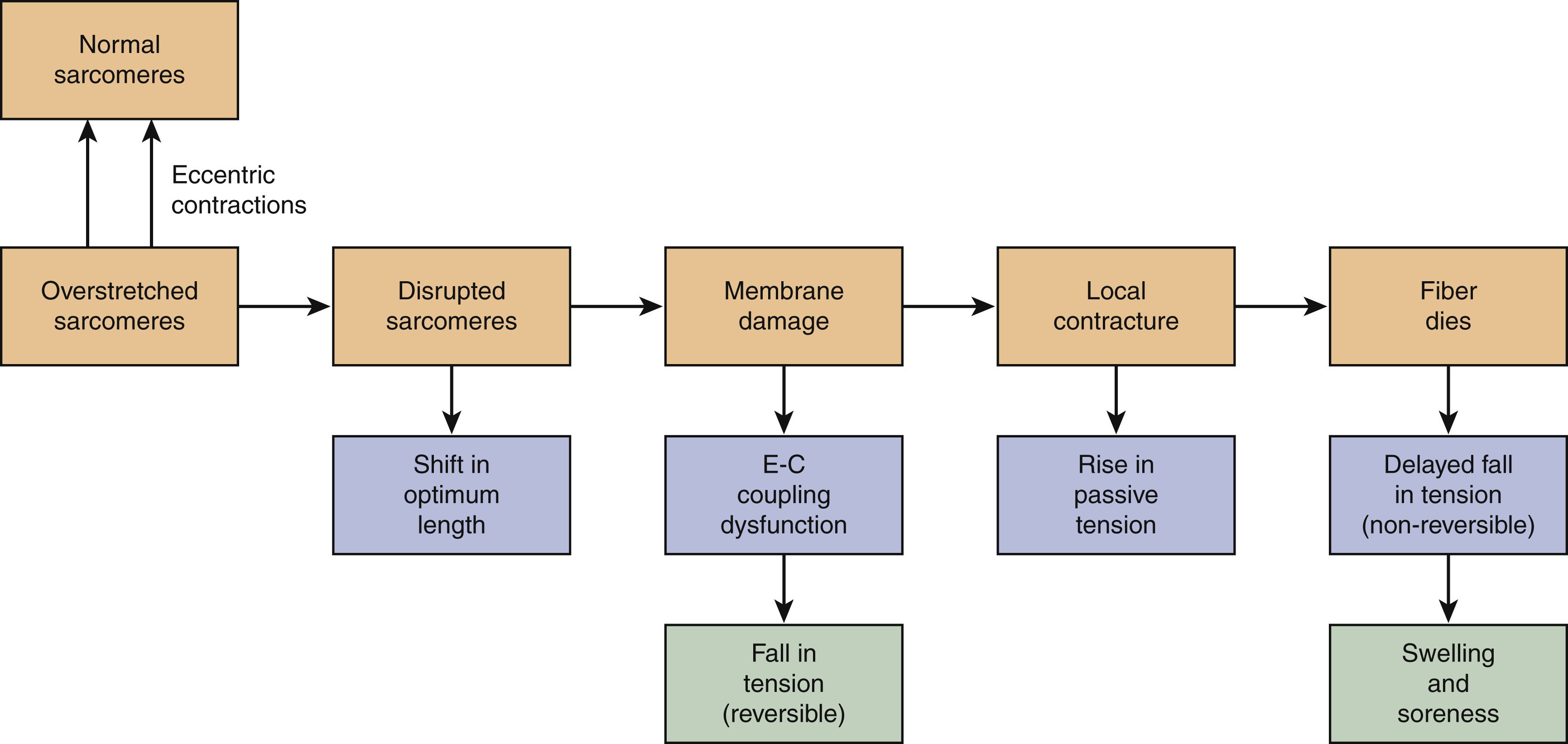
As this process begins to spread from a cellular level to a macroscopic level, specific facial muscles, such as the platysma, begin to develop contractures. This then creates a vicious cycle of increasing muscle stiffness (tension), reduced movements, and further contracture. The subsequent loss of the internal milieu of calcium homeostasis within the facial muscles hastens a process that is antagonistic to thixotrophy, called rheopexy. , At this stage, increasing passive stretches leads to further muscle stiffening, which is difficult to reverse.
Correlating this clinically, the consequence is that as the stiffness of facial expressions increases, more effort has to be put into the surrounding muscles to overcome this inherent resistance. Increased muscular effort is accompanied by recruitment of agonist muscles, a phenomenon called “motor overflow,” which manifests in facial muscles as “co-contractions.” As shown by Korb et al. in 2016 in a study of the normal population, increased effort when smiling, for example, concurrently activates other facial muscles such as orbicularis oculi and corrugator supercilii, as well as the masseter muscle. In simple terms, as it takes much more effort to overcome facial stiffness, more facial co-contractions are evident, as illustrated in Fig. 6.3 .
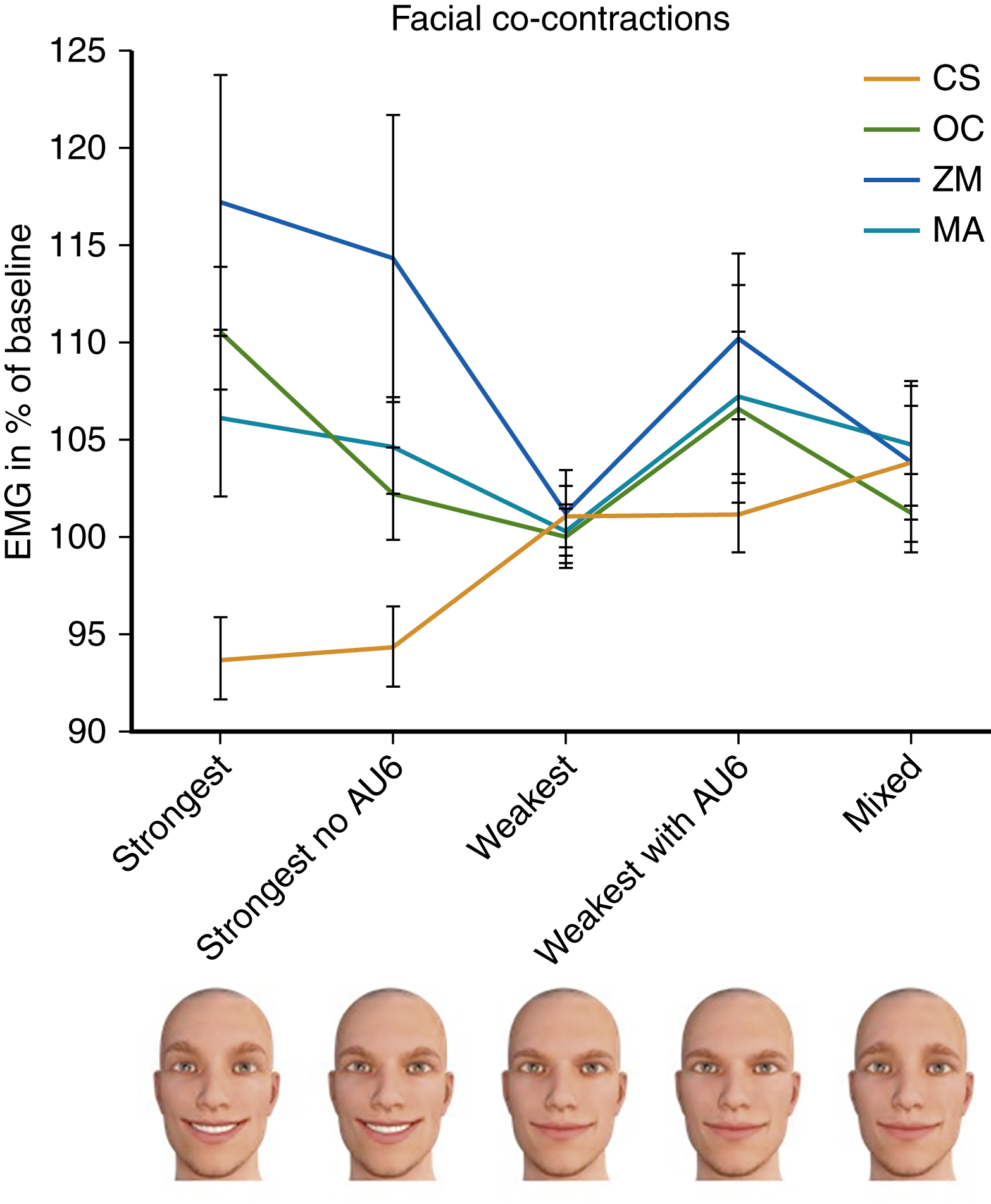
Note that corrugator supercilii tracings reduce on full smile activation in line with Sherrington’s law. This states that when one set of skeletal muscles is activated, e.g., the zygomaticus major, the antagonistic or opposing set of facial muscles, e.g., the platysma, is concurrently inhibited. Equally, the contralateral facial muscles should accordingly adjust as per Hering’s law. This has also been seen in the facial nerve.
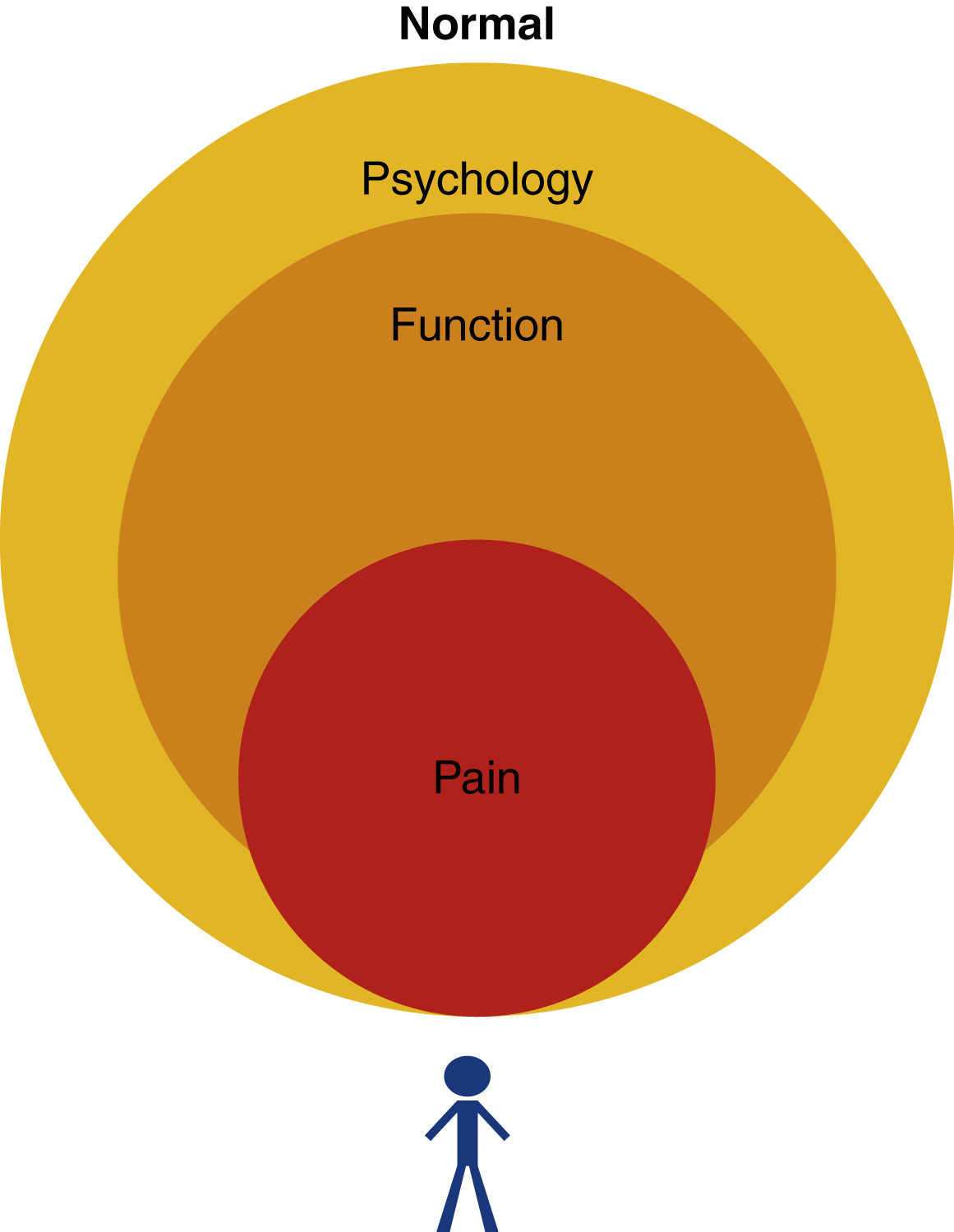
However, in faces afflicted by synkinesis, the issue is not only driven by the central nervous system (CNS) but rather due to local factors such as muscle stiffness and contracture. This complicates the typical agonist-antagonist muscle relationships leading to increased abnormal facial co-contractions (synkinesis). Hyperkinesis, which is excessive muscle activation, may affect the unaffected side and also contributes to this stalemate. This leads to a “locked face” situation, which comparatively increases the ratio of rheopectic to thixotropic muscle fibres, thereby causing more irreversible damage as alluded to in Fig. 6.2 . The key is to therefore break this vicious cycle before it reaches this stage and patients begin to develop functional impairment, reduced quality of life, altered psychology, and pain. Fig. 6.4 schematically represents the relationship between the inherent lack of proprioception, overactivity, tightness, pain, and functional/aesthetic sequelae.
Principles of Treatment
A variety of treatment options currently exist for non-flaccid facial palsy including facial therapy with biofeedback, chemo-denervation with botulinum toxin (BTX) and surgical interventions covered elsewhere in this book. In the author’s practice, we approach this from a pathophysiological standpoint with a view to reduce muscle stiffness. Key to this is embodied within Hill’s “Three-Element” Muscle Model ( Fig. 6.5 ), which explains that intrinsic muscle fiber stiffness is due to elements both “in-series” and “in-parallel.” The “in-series” element consists of two components – a contractile element (muscle) and an elastic component (sarcolemmal tube), whereas the “in-parallel” component refers to the elasticity of the surrounding soft tissue.
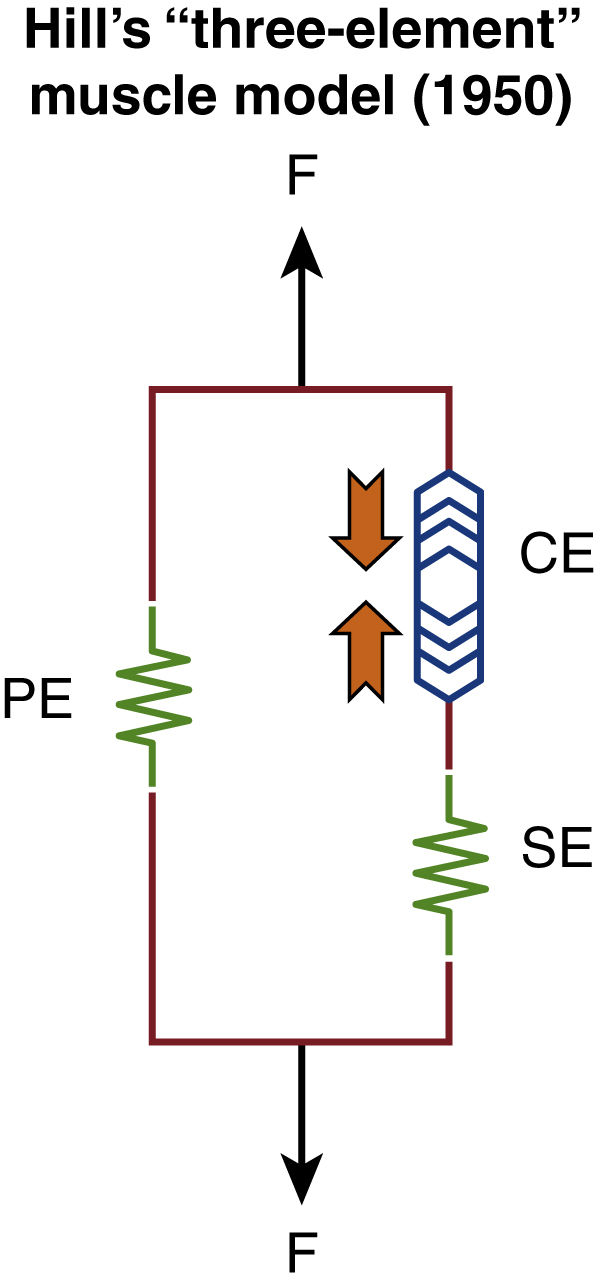
In the initial few months of facial reinnervation, typically seen in the first 6 months of Bell’s palsy, an increase in the number of actin-myosin cross-bridges causes gradual sarcolemmal shortening but this is mostly reversible in our experience if full reinnervation is returned within 3 months. These patients typically respond well to targeted facial therapy and avoiding over-exertion of the facial muscles and thereby eccentric contraction. In the East Grinstead Grade of Stiffness (EGGS) treatment-based classification, which we use in our practice ( Table 6.1 ), this is termed grade I, where the status quo of Hill’s muscle model is maintained.
| EGGS | Clinical Characteristic | Treatment |
|---|---|---|
| I | Facial stiffness which improves on stretching | Facial therapy only |
| II | Facial stiffness which partially improves with stretching and has reached a plateau point in symptomatic improvement | Facial therapy and targeted botulinum toxin-A (BTX-A) injections |
| III | Locked face with significant discomfort and pain | Selective myo-neurectomy |
If reinnervation returns at a slower rate and poorly coordinated muscle movements continue for longer periods, the proportion of cases with eccentric contractions increases, which in turn leads to sarcomere damage. Within Hill’s model, this leads to reduced “give” within the system and further stress on the elasticity of the sarcomere, giving rise to a thixotropic state (EGGS II). Although some of the passive tension will reduce with stretches and relaxation exercises, low-dose BTX injections will deactivate some of the actin-myosin bridges, thereby also contributing to an overall decrease in passive tension and thereby less sarcomere damage. The combination of facial therapy and BTX therefore can mitigate against the effect of thixotrophy as shown in Fig. 6.6 .
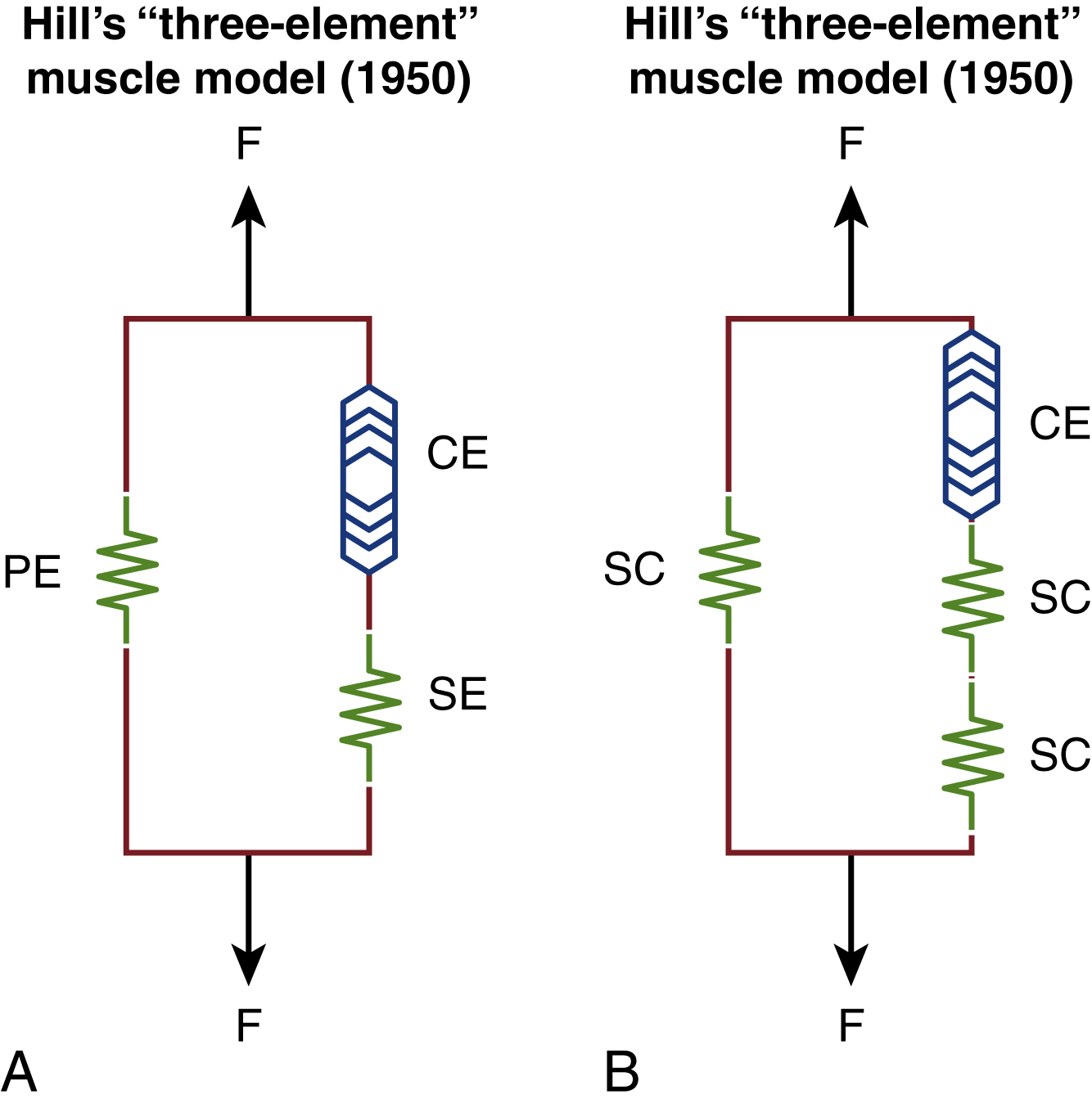
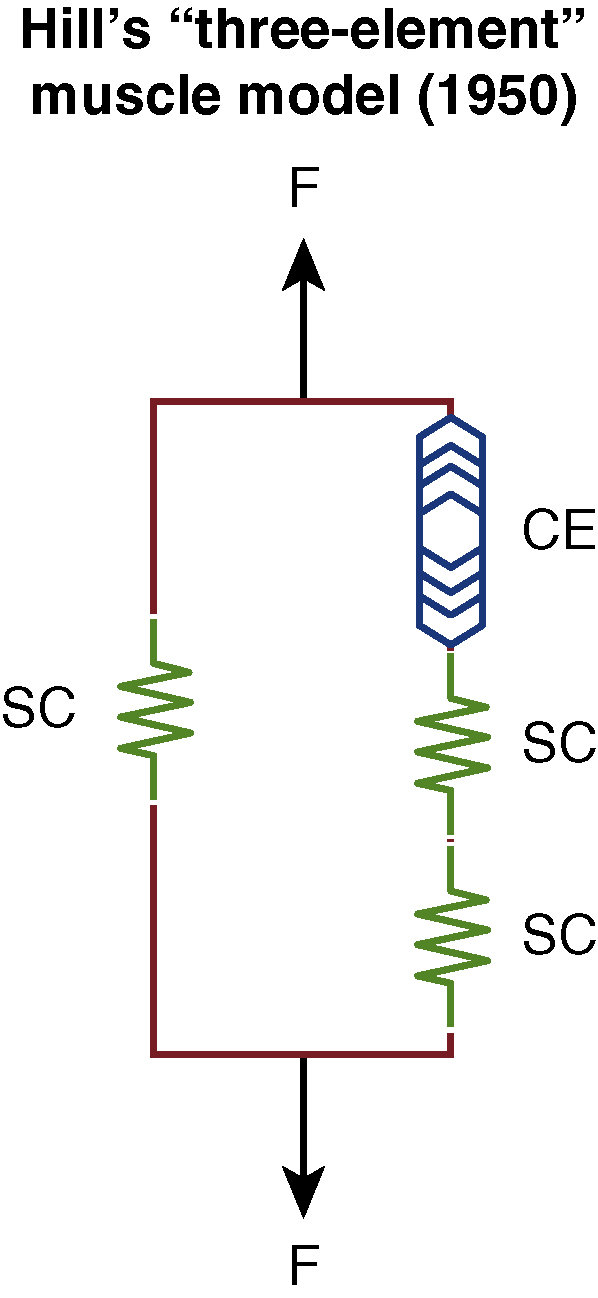
However, if these patients present typically beyond 2 years and the vicious cycle of tension and sarcomere damage continues unabated, muscle fibers then fibrose, thereby causing contractures (EGGS III). At this stage, as a greater proportion of the affected muscle contracts, rheopexy begins to set in instead and patients then typically present with discomfort and even pain in the most severely affected areas as there is entrapment of sensory nerves. As shown in Fig. 6.7 , almost all of the muscle in this instance is “inelastic” fibrotic tissue where only surgical release procedures, e.g., selective neurectomies, can help in certain areas such as the neck (platysma).
Botulinum Toxin
Botulinum toxin-A (BTX-A) is one of the most potent toxins known to man. Injections of BTX-A are useful adjunct in the treatment of non-flaccid facial palsy, having been used for this purpose since 1987. Clinically it reduces the over-activity of muscles on the ipsilateral, affected side, while reducing unnecessary force from the contralateral side, to minimize the overall risk of eccentric contractions which, alongside coordinated facial therapy, helps to reverse the effects of thixotrophy.
BTX-A is derived from an anaerobic gram-positive bacillus, Clostridium botulinum , which has many subtypes, viz. A to G. However, in clinical practice it is BTX-A that is commonly used, of which there are three versions: onabotulinumtoxinA, incobotulinumtoxinA, and abobotulinumtoxinA. Commercial preparations of these exist as Botox, Xeomin, and Dysport, respectively. These have similar active moieties but differing molecular structures and hence, diffusing capacity. Each has different diluting strengths prior to use.
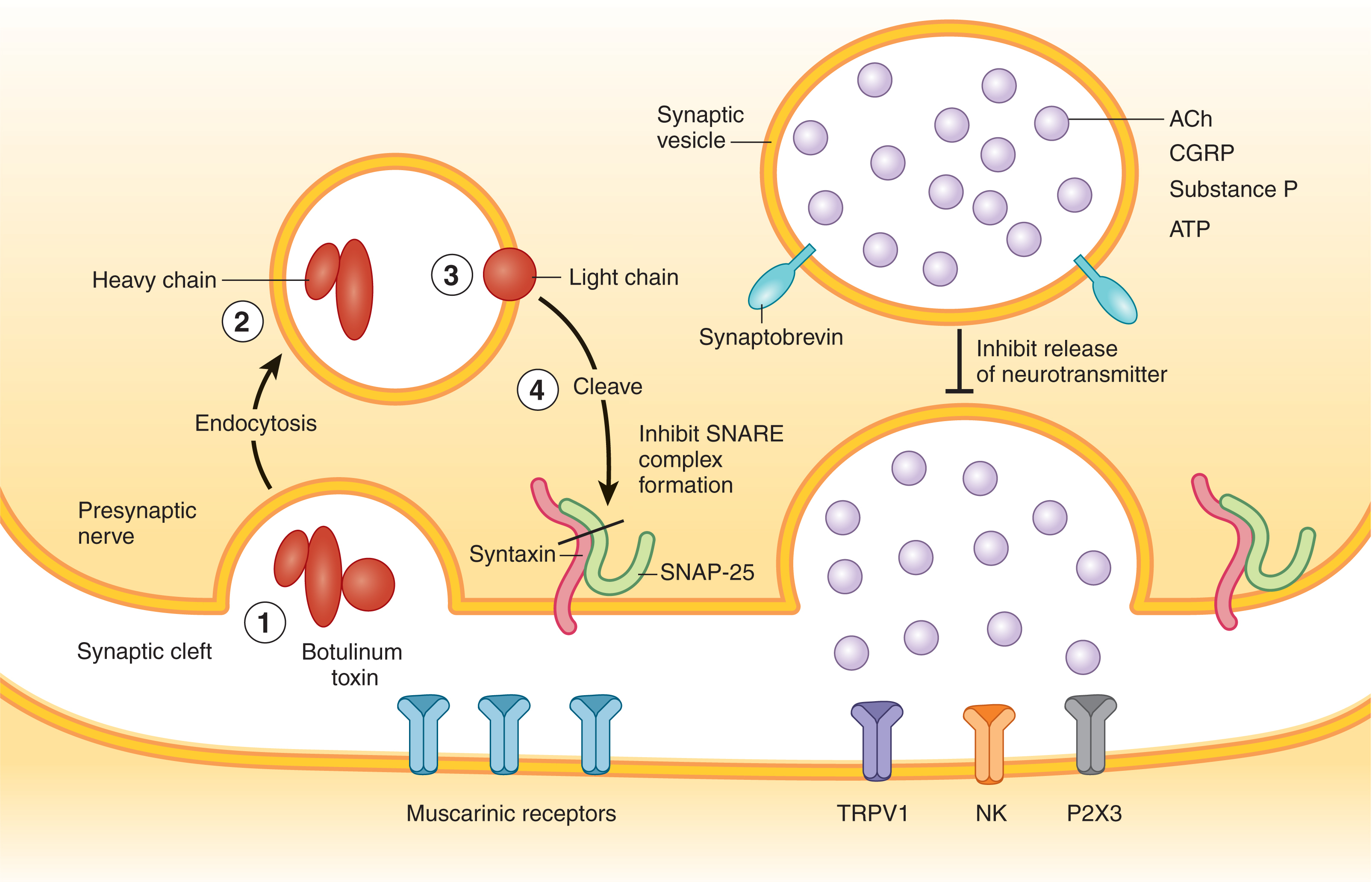
BTX-A works in the same fashion whatever its trade name ( Fig. 6.8 ). BTX is taken up at the level of the neuromuscular junction (NMJ) by a process of endocytosis and within these vesicles, it is cleaved into its constituent heavy and light chains, the latter of which is the active moiety, a 150 kDa protein. This protein then attaches to SNAP-25, a protein molecule which in turn deactivates synaptobrevin, thereby inhibiting acetylcholine (Ach) release for the continuation of saltatory conduction. These effects typically start as early as 3 to 14 days in patients and can last from 4 to 6 months.
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


