Fig. 3.1
Partitioning of daily energy expenditure (EE). Resting energy expenditure (REE) represents 65 % of daily EE, thermic effect of food (TEF) or food thermogenesis 10 %, and physical activity (PA) the remaining 25 %
3.2 Physiology of Caloric Intake and Energy Expenditure
The basic physiological mechanisms that regulate the energy balance in the central nervous system (CNS) derive from the afferent signals from the periphery regarding satiety and adiposity, which trigger efferent neuro-hormonal activation aimed at reducing appetite and maintaining energy balance [3, 4].
Afferent signals derive from the gastrointestinal tract, including pancreas and liver, in the form of autonomic stimulation by physical and chemical food ingestion [5]. Even more importantly, a series of hormones are released by the gut which assist in nutrient digestion and absorption and regulate through the vagus satiety feeling. Among them there are cholecystokinine (CCK) which stimulates exocrine pancreas function, peptide YY (PYY) which increases energy expenditure and slows gastric emptying as well as it is done by glucagon-like peptide 1 (GLP1) which, in addition, has a potent effect on endocrine pancreatic function [6, 7]. Finally, insulin, glucagon and amylin, beside their effects on carbohydrates metabolism, act on the hypothalamus stimulating satiety. On the contrary, ghrelin, a potent orexigenic peptide, secreted by the fundus of the stomach when empty, is suppressed by food ingestion. The other type of afferent signals regulating food intake, comes from the adipose tissue which is not only a fat storage compartment but, as it has been progressively recognized, plays an active role in maintaining energy balance. In fact, the adipose organ produces a number of mediators with important physiological functions and impacts in the development of diseases linked to obesity [8–10].
More than 50 adipose tissue hormones and cytokines have been described including acute phase proteins, complement-like factors and adhesion molecules. These so-called adipokines are involved in the regulation of satiety, energy balance, lipid and glucose metabolism, and inflammation [11]. There are two types of adipose tissues, the “white” and the “brown” [12]. The former is prevalent in mammals and is made of round adipocytes containing a large fat droplet which displaces the small nucleus to the periphery. The brown adipocyte is smaller with many small fat accumulations and is rich of mitochondria which are responsible for the darker appearance. Thermogenesis is the primary function of this type of adipose tissue and, in fact, contrary to the white one, it contains uncoupling proteins which prevent storage of energy in the ATP molecules. It is also highly vascularized and innervated by sympathetic fibers. On the other hand, the white adipose tissue, which is widely prevailing in the adult, beside adipocyte, contains macrophages, fibroblasts, leukocytes, and endothelial cells which are responsible for the secretion of additional mediators to those peculiar of adipose tissue. The most well known adipokines are leptin, adiponectin, tumor necrosis factor α (TNFα), interleukin 6 (IL6), and resistin.
Leptin is a peptide with a central function in the regulation of body weight through limitation of food intake and enhancement of energy expenditure. Being produced by adipocytes, its blood level is directly proportional to the fat mass. In turn, leptin binds to the leptin receptor in the hypothalamus with the activation of anorexigenic response. Leptin and its receptor are regulated by separate genes whose mutation leads to endocrine abnormalities including obesity similar to the one observed in the ob/ob mice, deficient in leptin, and in the db/db mice that have a deficit of leptin receptors [13]. The relative interplay between leptin production and leptin receptor expression may partly explain the so-called leptin resistance phenomenon which is commonly observed in patients who are obese in the face of high circulating levels of leptin [14]. Another very interesting action of leptin, beside the regulation of appetite and of energy expenditure, is a direct stimulation of T lymphocytes with increased inflammatory cytokines production such as TNFα and IL6.
Adiponectin is a cytokine-like molecule that interacts with its specific receptor on the cell membrane of the central nervous system, of the muscles and of the liver. The action of the adiponectin is antithetic to leptin being reduced in obesity, increasing glucose and fatty acids uptake and reducing production of TNFα and IL6 and of other inflammatory mediators [15].
Resistin is a protein which is secreted mainly by adipocytes and macrophages, and owes its name to its enhancing effect on insulin resistance. This might be due, at least partially, to the increased hepatic glucose production and to the impaired glucose uptake and glycogen synthesis effect. In addition, it displays an inflammatory action especially on smooth muscle cells through stimulation of the immune system [16].
3.3 Central Regulation of Energy Homeostasis and Efferent Signaling
The central nervous system is the site where converge afferent nervous and hormonal signals so far described, providing information on satiety and adiposity and where effector neuro-chemical signals are activated in order to maintain the energy balance.
The appetite brain network includes the insula, the amygdala/hippocampus, the orbitofrontal cortex with the ventromedial prefrontal cortex and the nucleus striatum (Fig. 3.2). Relevant information, in particular sight and smell, reaches the brain as external cues acting on the amygdala and insula; interoceptive information from the gut is sensed by the insula, while circulating peptides and nutrients are sensed by the hypothalamus and brainstem, as well as by the ventral tegmental area and the substantia nigra. The amygdala encodes the current incentive value of food cues, while the insula conveys sensory features of foods and its activity is modulated by hunger. The cognitive control over appetite regions, either to enhance or to suppress appetite, is mediated mainly via the frontal lobes. Fasting increases activation of the hypothalamus, insula, and striatum, while meal consumption increases activation of the prefrontal cortex. A recent review [17] has highlighted how obesity is consistently associated with heightened or abnormal responses to visual food cues in a distributed network of brain regions involved in reward/motivation and emotion/memory and how a prolonged long-term exposure to highly palatable, high-calorie foods may cause decreased reward area activation following food intake. By using a positron emission tomography (PET) approach, Del Parigi et al. [18] found that obese individuals showed a greater activation in the midbrain and middle-dorsal insula that are areas involved in the cerebral reward response, and a lesser activation in the posterior cingulate cortex, which is associated with awareness state. PET studies demonstrated that obese adults show a lower postprandial activation of the cognitive control areas than lean individuals. EEG studies described the temporal evolution of the brain responses to face and food pictures, highlighted differences in resting state cortical networks among underweight, normal-weight, and overweight/obese subjects and changes in responses of the obese subjects with respect to the normal-weight individuals.
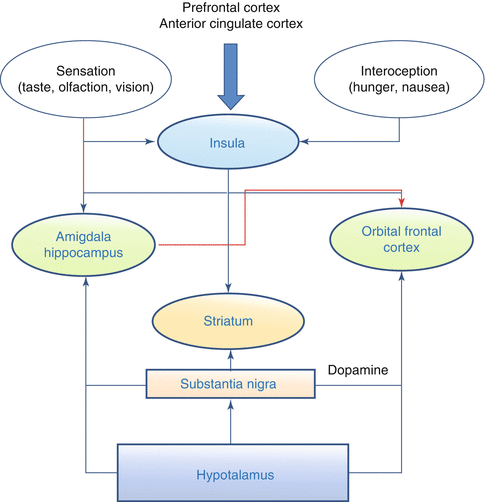
Fig. 3.2
Appetite Brain Network. Information from the prefrontal cortex and the anterior cingulated cortex arrive to the insula, which receives also afferences relative to food sensations like taste, olfaction and vision, and interoceptive sensation, such as hunger. Efferences from the insula reach the amigdala and hippocampus and the orbital frontal cortex and the striatum. The latter has also afferences from the substantia nigra that reach also the orbital frontal cortex and the striatum. Finally, the hypothalamus sends information to the substantia nigra, the orbital frontal cortex, and the striatum. In red are highlightened the inhibitory actions
In the arcuate nucleus, there are two types of neurons [19]: the first with anorexigenic and the second with orexigenic effects. The first type produces pro-opiomelanocortin peptide which interacting with the ventromedial as well as the other hypothalamic nuclei reduces food intake and increases energy expenditure. On the contrary, the orexigenic neurons synthesize PYY and aguti-related peptide which increase food intake and reduce energy consumption (Fig. 3.3). The hypothalamus receives sensory satiety signals from the peripheral organs through the vagal complex in the hindbrain and also directly from the blood carrying nutrients and peptides from the digestive system as well as from the adipose tissue. Finally, additional signal are acquired from limbic and cortical areas regarding cognitive and reward response to food intake [4].
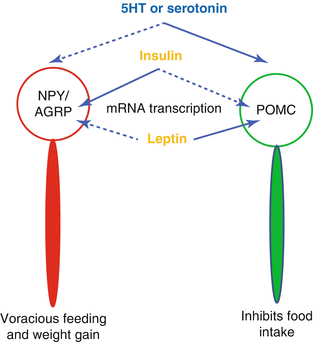
Fig. 3.3
Stimulatory effects are indicated by solid lines and inhibitory effects by dashed lines. 5-hydroxy-tryptamine or serotonine as well as leptin stimulates pro-opiomelanocortin (POMC) neurons and inhibits the neuropetide Y(NPY)/agouti related protein neurons. Insulin does the contrary. Therefore, insulin stimulates appetite while serotonin and leptin do the opposite
3.4 Pathophysiology of Obesity
3.4.1 Environmental and Genetic Factors
Regardless of whether obesity is a condition or a disease, it arises from multiple etiologic determinants, either inherited or acquired. In particular, the strict interplay of environmental and genetic factors is strongly correlated to obesity development. Environment contributes providing and advertising easy access to calorie-dense and palatable food, facilitating sedentary lifestyle, while increasing prevalence of mental distress or illness either by itself or because of related medications [20]. It represents an important determinant of the obesity epidemics especially in the western countries. Environmental factors act on individual genetic background which animal as well as human family studies have shown to be a major determinant of obesity. Indeed, weight is a highly inheritable trait although greatly polymorphic and polygenic. More than 40 genes have been so far linked to obesity which, interacting with the environment, may result in its relevant phenotypic expression [21]. At the present time, however, carrying obesity genes have little recognizable effect and, therefore, should be considered as more of a risk factor than a determinant [22]. Thus, an epigenetic component in the pathophysiology of obesity should be taken into consideration. In fact, recent works have shown how genome mutations, that do not involve changes in DNA sequence, could explain how environmental conditions, like maternal food intake during pregnancy and overfeeding during infancy have an impact on obesity susceptibility [23, 24]. However, there are some monogenic obesity syndromes with well characterized single locus mutations especially in the pediatric population. Among them it should be mentioned the Prader-Willis syndrome (in which seven genes of paternal origin are deleted), the Bordet-Biedl syndrome, the leptin deficiency syndrome, and other rare diseases all associated with complex neurological abnormalities.
3.4.2 Obesity as Derangement of Central Regulation of Energy Balance
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


