Gastricbanding (%)
Gastroplasty(%)
Gastricbypass (%)
Biliopancreatic division/duodenal switch (%)
Total (%)
% excess weight loss
−47.45
−68.17
−61.56
−70.12
−61.23
Resolution type2 diabetes
47.9
71.6
83.7
98.9
76.8
Resolutionhyperlipidemia
58.9
73.6
96.9
99.1
79.3
Resolutionhypertension
43.2
69.0
67.5
83.4
61.7
This volume is primarily concerned with the metabolic surgery procedures of bariatric surgery, which, in turn, have given rise to metabolic operations specifically designed to mitigate certain of the comorbidities of obesity, with or without weight loss, in particular type 2 diabetes. This chapter will, therefore, discuss the old and newly proposed mechanisms of action for these procedures, the specific neurologic networks and hormones involved, the energy metabolism of obesity, the involvement of inflammation, and the present and future outcomes for this acquired knowledge.
8.1 Mechanisms of Action
Traditionally, the weight loss achieved by bariatric surgery has been attributed to either a restriction of food intake or malabsorption of unrestricted oral consumption. This simplistic approach, of course, ignores the minimal weight loss success achieved by electronic stimulation procedures [2, 3, 16] that employ no overt restriction of eating or of absorption. In the final analysis, at the level of caloric absorption from the intestinal tract, all the common bariatric surgery procedures are restrictive. This principle is illustrated in Fig. 8.1a of the steps in ostensibly restrictive bariatric surgery and in Fig. 8.1b of the steps in ostensibly malabsorptive bariatric surgery. Caloric intake can be restricted by the inhibition of eating caused by a gastric balloon, a gastric band, a sleeve gastrectomy, or a gastric bypass. Caloric intake can be equally restricted by the insufficient intestinal absorptive surface available in the so-called malabsorptive procedures. Since the bariatric surgery operations, even those with the ultrashort common channels of a biliopancreatic diversion or duodenal switch, extremely rarely cause excessive weight loss, there must be a brake effect in body metabolism not explainable by the old mechanisms of action concepts. Further, there are many nonobese, even lean, individuals who have undergone gastric or massive intestinal resections for various reasons and who subsequently maintained their body weight. It is time, therefore, in order to understand the changes we are eliciting by metabolic bariatric surgery, that we acknowledge that we do restrict the ability to eat and that we do restrict body caloric intake, but that we must look to more sophisticated explanations for the true mechanisms of action of these procedures. We need to investigate the complex neurohormonal networks that control weight, certain weight-related diseases, and the disease of obesity itself.
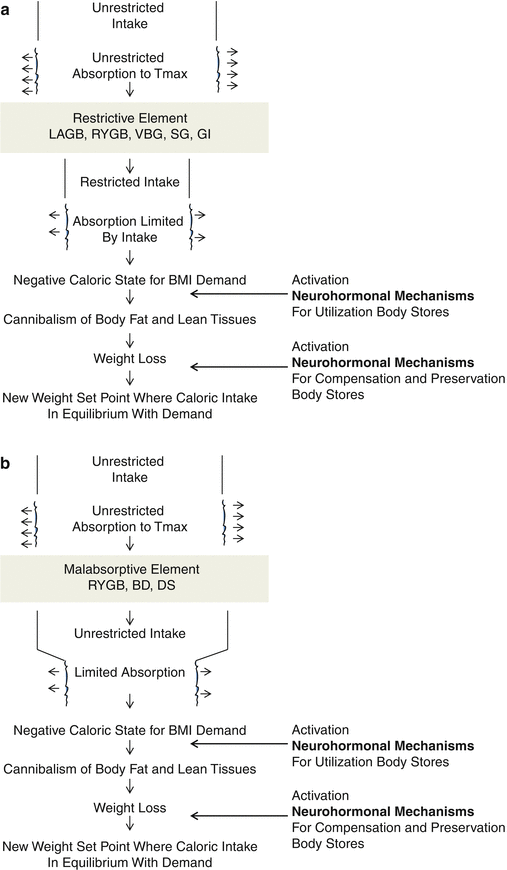
Fig. 8.1
Traditional explanatory mechanisms for bariatric operations: (a) Restrictive, (b) malabsorptive. Key: LAGB laparoscopic adjustable gastric band, RYGB Roux-en-Y gastric bypass, VBG vertical banded gastroplasty, SG sleeve gastrectomy, GI gastric imbrication, BPD biliopancreatic diversion, DS duodenal switch
8.1.1 Neural Networks
There exists vigorous parasympathetic and sympathetic innervation to the intestinal tract. Old and extensive teaching of vagal nerve function concentrated on efferent stimulation of acid/pepsinogen secretion by the stomach, control of bile production and gallbladder function, and output of pancreatic exocrine enzymes [26, 27]. However, only 10–20 % of vagal fibers are efferent; 80 % or more are afferent. We know that these afferent connections go to the brain, primarily to the hypothalamus and medulla oblongata [28]. These fibers are intimately associated with hypothalamic functional centers related to hunger, appetite (post-hunger satisfaction eating behavior), and satiety [29]. Prosthetic gastric volume restrictors, gastric imbrication, adjustable gastric bands, Roux-en-Y gastric bypass, biliopancreatic diversion/duodenal switch, and simple sleeve gastrectomy, as well as the experimental procedures of gastric and vagal stimulation, duodeno-jejunal bypass, and endoluminal sleeves, all influence vagal, in particular afferent vagal, nerve function.
Sympathetic gut innervation is primarily mediated via the celiac plexus. Efferent sympathetic fibers are involved in glucose production and release, inhibition of gastrointestinal enzymes, and inhibition of peristalsis [30]; afferent sympathetic fibers are traceable to cerebral pain receptors. Roux-en-Y gastric bypass, biliopancreatic diversion/duodenal switch, and certain of the other metabolic/bariatric procedures, can be expected to induce a sympathetic neurologic response.
There is also an intrinsic nerve syncytium, primarily in the submucosal layer of the intestinal tract, extending from the esophagus to the anus [31] (Fig. 8.2). Surely, this network is altered by many of the metabolic/bariatric operations, for example, Roux-en-Y gastric bypass, biliopancreatic diversion/duodenal switch, and ileal transposition. The density of the submucosal neural fibers has been demonstrated to be an indicator of type 2 diabetes severity [32]. Finally, with respect to the participation of intestinal neurogenic pathways in the mechanisms of action of the metabolic/bariatric operations, there exists the high fundic gastric pacemaker [33], which is disconnected by a Roux-en-Y gastric bypass and biliopancreatic diversion, and totally extirpated by sleeve gastrectomy and duodenal switch.
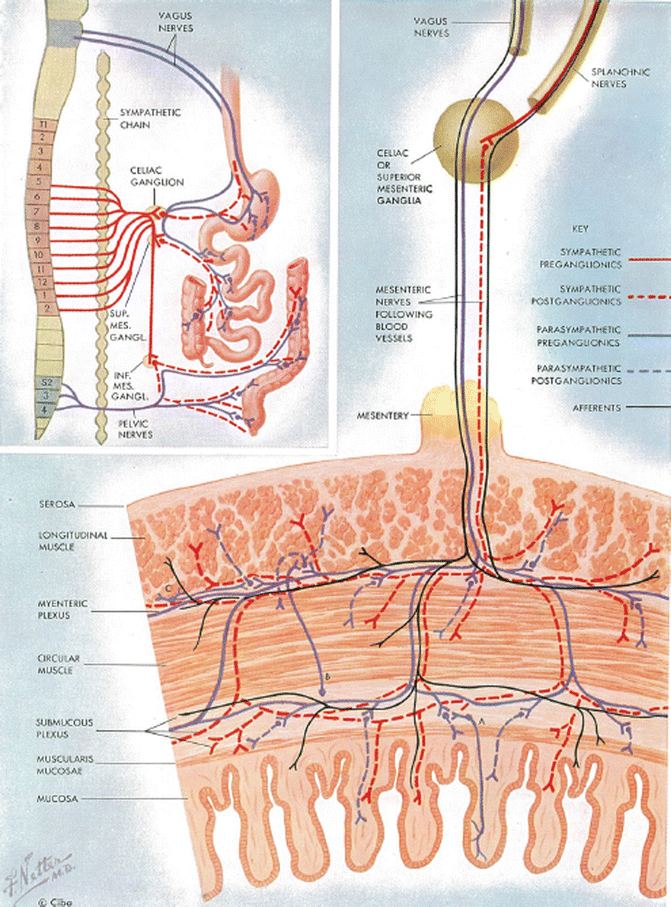
Fig. 8.2
Vagal and sympathetic innervation of the stomach and intestine; intestinal wall nerve syncytium (Reprinted from Netter [31], with permission from Elsevier)
8.1.2 Gut and Fat Derived Hormones
Complementing the neural network in influencing weight and obesity control mechanisms, as well as intimately involved in the obesity comorbidity of type 2 diabetes, are the gut and the fat derived hormones. Over the course of time, more and more individual intestinally and fat secreted hormones have and are being identified. Currently, there are about 100 of these unique peptides and they each have one or several functions [34]. Due to the concentrated efforts of dedicated investigators, the role and significance of specific gut hormones in the responsible mechanisms for the metabolic/bariatric procedures have been emphasized. Over time, the popularity of these individual hormones has risen and fallen, and possibly risen again. The true importance of each documented hormone in obesity and type 2 diabetes is as yet not precise. It is also likely that mechanisms of action are never dependent on a single hormone but rather on a mosaic of hormonal interactions. It is, therefore, pertinent to review the actions and properties of certain of the better understood hormones. Primary interest by metabolic/bariatric surgeons on hormonal mechanisms has focused on glucagon-like peptide-1 (GLP-1), peptide YY (PYY), leptin, ghrelin, and glucose-dependent insulinotropic peptide (formerly gastric inhibitory peptide) (GIP).
GLP-1 and PYY are both elaborated by the L-cells of the intestinal mucosa and seem to work in concert, eliciting the same metabolic responses or augmenting the actions of the other. Their gastrointestinal properties include the reduction of hunger and an increase in the sensation of satiety, resulting from stimulation of the arcuate nucleus of the hypothalamus [35–37]. They are also responsible for the “ileal brake” effect of delayed gastric emptying, delayed mouth-to-cecum transit time, and decreased jejunal wave pressure [38–40]. Regarding pancreatic endocrine function, GLP-1 and PYY contribute significantly to the incretin effect, defined as the concentration of insulin release by oral glucose stimulation that exceeds the insulin concentration elicited by the same amount of intravenous glucose [41]. The influence of GLP-1 on pancreatic endocrine function is direct and appears to be more powerful than that of PYY. GLP-1 stimulates glucose-dependent insulin secretion, preinsulin gene expression, β-cell proliferation and antiapoptotic pathways, and inhibits glucagon release [42, 43]. It has been shown that GLP-1 secretion is reduced in patients with type 2 diabetes [44, 45], which may be responsible for the hyperglycemia of this disease [46]. The PYY effect on insulin secretion is indirect by inhibiting the action of gastrin-stimulating peptide and gastrin-releasing peptide [47, 48].
It is interesting that PYY exhibits two active circulating forms: PYY (1-36) and PYY (3-36). PYY (1-36) increases appetite and promotes weight gain, and is not the PYY hormone that interests metabolic/bariatric surgeons. PYY (3-36) is the form cited in metabolic mechanisms literature and is responsible for centrally decreasing appetite and promoting weight loss [49–53]. PYY (3-36) also inhibits pentagastrine-stimulated gastric acid secretion [54] and the cephalic phase of pancreatic exocrine secretion [55, 56].
Both GLP-1 and PYY are secreted in increasing amounts as a function of the caloric content of ingested food [57]. Other influencing factors include the intestinal site of stimulation [58–60], bile acids [61], central neural and vagal mechanisms [62–64], and other hormones (vasoactive intestinal peptide [65], gastrin [66]). Leptin forms a triad with GLP-1 and PYY in weight regulating mechanisms, possibly providing for an autoregulatory feedback loop. Leptin stimulates GLP-1 secretion and GLP-1 suppresses leptin levels [67].
Leptin, a peptide hormone synthesized and released by adipocytes, has been implicated, by its action on mediobasal hypothalamic receptors, in maintaining a body weight set point [68–70]. When fat mass falls, plasma leptin levels fall as well, stimulating appetite and suppressing energy expenditure; when fat mass increases, plasma leptin levels increase, suppressing appetite and increasing energy expenditure [71–73]. Thus, apart from its reciprocal autoregulation mechanism with GLP-1, leptin’s specific hypothalamic effect on suppressing eating is parallel to that of GLP-1. Leptin’s relationship to obesity, however, is far from clear cut. Much of the leptin physiologic data were derived from rodent models and have not been equivalently substantiated in humans. Obese individuals generally have a high, not low, circulating concentration of leptin [73] and are stated to be leptin-resistant [71]. It is apparent, therefore, that the normal leptin feedback control in obese humans is flawed.
In addition to its action on the circulatory system, lung surfactant activity, bone, reproduction, and areas of the brain not involved in weight regulation, leptin is an inflammatory marker responding specifically to adipose-derived cytokines [74–77]. Increases in leptin levels in response to overeating may, therefore, play a role in inflammation-related diseases, including not only obesity but diabetes, hypertension, and cardiovascular disease.
Ghrelin is a peptide hormone produced by P/D1 cells in the fundus of the stomach and the episilon cells of the pancreas [78]. There are ghrelin receptors in the pituitary, stomach, intestine, pancreas, thymus, gonads, and heart [79]. Some of ghrelin’s actions may be due to its potent stimulation of growth hormone secretion from the anterior pituitary [80]. Most interestingly, ghrelin selectively reduces mechanosensitivity of upper gastrointestinal vagal afferents [81]. Ghrelin’s actions are in opposition to those of leptin: it increases hunger and eating [82, 83]. Circulating ghrelin levels are the highest before a meal and at their lowest thereafter [84]. Intravenously administered ghrelin increases food intake in a dose-dependent manner [85]. However, plasma ghrelin levels in the obese are lower than in lean individuals [86], suggesting that ghrelin does not contribute to obesity. An exception is Prader–Willi Syndrome where ghrelin levels are high [87, 88]. Ghrelin appears to be an anti-inflammatory hormone in opposition to the cytokine properties of leptin [89, 90].
GIP, secreted by the K-cells of the duodenum, along with GLP-1, is an incretin hormone [91]. In the past, it was believed that GIP neutralized and inhibited stomach acid, decreased gastric transit time, and retarded intestinal motility; however, recent information attributes these actions to secretin and not to physiologic levels of GIP. Thus, GIP is really not a gastric inhibitory hormone. It is now believed that the primary function of GIP is to induce insulin secretion via glucose hyperosmolarity in the duodenum [92]. Hence, a name change to glucose-dependent insulinotropic peptide, allowing for retention of the acronym “GIP.” Type 2 diabetics are not responsive to GIP administration and have lower postprandial levels of GIP secretion compared to nondiabetics [93].
As our knowledge base evolves, other hormones will arouse interest and controversy with respect to their effects on normal body weight, obesity, type 2 diabetes, and other comorbidities of obesity. At present, attention is being paid to pancreatic polypeptide, cholecystokinin, oxyntomodulin, and adipose-derived adiponectin and resistin.
8.2 Energy Metabolism of Obesity
Having defined metabolic surgery, we should now more closely examine the energy metabolism of obesity, the primary working domain of metabolic/bariatric surgery. Past knowledge of obesity causation and mechanisms of action focused on genetics and energy balance [94, 95]. The genetics of obesity is not truly a subject to be covered under metabolic surgery or metabolics; however, energy balance is appropriately considered under metabolic mechanisms.
Total body metabolic processes in kinetic terms can be defined as total energy expenditure (TEE), which is equal to the basal metabolic rate (BMR), the energy of activity expenditure, the thermal effect of food (TEF), and adaptive thermogenesis (AT). The BMR is the lowest level of energy needed to maintain life (e.g., autonomic cardiorespiratory functions). The TEF is the body heat produced and the AT is the fluctuation in TEF produced by environmental temperature and humidity [95]. The homeostatic responses to increased food intake in normal individuals essentially is an attempt to resist weight gain; whereas, in obese individuals these mechanisms appear to be blunted resulting in a decrease in the TEF and a subsequent decrease in the TEE [96].
The nature of energy metabolism after metabolic/bariatric surgery has not been extensively studied and is not well understood. The preoperative TEE of patients undergoing gastric bypass can vary from normal to hypermetabolic. In the former, there appears to be no change postoperatively; in the latter, normalization of TEE seems to occur postoperatively [97]. Further, preservation of energy economy is reciprocal to the degree of weight loss [98]. After gastric bypass, both TEE and resting energy expenditure are decreased by about 25 % [99].
8.3 Inflammation
Currently, underlying causative mechanisms for obesity and type 2 diabetes are often focused on the state of inflammation. An increase in energy (caloric) intake over energy expenditure leads to an increase in the size of the adipocytes. Once the adipocyte volume exceeds 1 μl of lipid, the number of fat cells increases. The ensuing leptin response in non-obese individuals reduces hunger and downregulates caloric intake. In the obese, with a blunted leptin response [71], the unchecked caloric load overtaxes the adipocytes’ ability to grow larger or increase in number resulting in a state of inflammation at the cellular level and ectopic fat storage in internal organs, including the liver and muscle [76]. In addition, the consumption of high volumes of fructose, absent in our ancestral diets, has been suggested to cause leptin resistance and elevated triglycerides, which result in weight gain [72, 100, 101] and a state of generalized inflammation.
The inflammation rationale carries over to obesity-induced type 2 diabetes. The increased fat cell mass results in an increase in free fatty acids, which, in turn, reduce glucose utilization by the liver and peripheral tissues, thereby reducing insulin clearance by the liver and increasing insulin delivery to the periphery, resulting in peripheral insulin resistance. Adipocyte-secreted cytokines contribute to the inflammatory state as well. The reactive oxygen species engendered by the inflammatory process are then responsible for endothelial damage, impaired beta cell function and, in general, play a cardinal role in the long-term complications of type 2 diabetes [102].
8.4 Conclusion
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


