Chapter 18 The pathophysiology of inhalation injury
 Access the complete reference list online at http://www.expertconsult.com
Access the complete reference list online at http://www.expertconsult.com
Introduction and epidemiology
It has been over two decades since the authors published their first manuscript on inhalation injury.1 In the review article published the next year, it was reported that inhalation injury was a main factor responsible for mortality in thermally injured patients.2 Inhalation injury is still a major problem.3 Although sepsis may be the major cause of death among burned children, two-thirds of the patients who have died in our own hospital have had an inhalation injury.4 Smoke inhalation causes 5000–10 000 deaths annually in the United States and more than 23 000 injuries, including approximately 5000 firefighter injuries.5 In fact, the United States has one of the highest fire death rates among industrialized countries. Inhalation injury is a serious medical problem. More than 30% of thermally injured patients admitted to burn centers in the United States have a concomitant smoke inhalation injury.6 Similar percentages of fire victims who have sustained smoke inhalation appear in several other countries.7–10 Despite effective management of fluid resuscitation, early surgical excision of burned tissue and improved ventilation techniques, the mortality rate of patients who have combined burn and smoke inhalation injury is still high.6,11–13 In patients with combined injury, the lung is a critical organ and the progressive respiratory failure associated with pulmonary edema is a pivotal determinant of mortality.14–16 Albeit not as lethal, smoke inhalation alone is a serious problem. It is estimated by the World Health Organization that over one billion people develop airway and pulmonary inflammation as a result of inhaling smoke from indoor cooking fires, forest fires and burning of crops (Fig. 18.1).17,18

Figure 18.1 An example of direct exposure to smoke as a result of open fire using various materials as a fuel.
The inhalation of toxic materials has been of interest for a number of years, especially as the result of their use in gas warfare. In the 1940s two very large fires focused interest on the inhalation of smoke in fire victims. The first was a fire at a nightclub in Boston called the Cocoanut Grove, where a large number of people were trapped in a burning building and consequently sustained severe inhalation injury.19,20 It is interesting that in recent times a similar fire occurred in a nightclub near Boston in Rhode Island.21 The second disaster occurred in 1947 in Texas City, Texas.22 Here, a ship loaded with ammonium nitrate fertilizer exploded in the harbor and set off a chain of explosions and fires among some 50 refineries and chemical plants, resulting in over 2000 hospital admissions of patients with burn injury, many of whom who had simultaneously inhaled smoke, as well as victims with smoke inhalation alone. In many ways the burn victims of the 9/11 disaster at the Pentagon were similar to these individuals, as the burns and inhalation involved combustion of petroleum products. Among the 790 injured survivors of the terrorist attack on the Word Trade Center in New York on 11 September 2001, 49% suffered from inhalation injury. The situation was the same as the attack on the Pentagon, and in both situations inhalation injury was also seen in some patients who were not burned.21–23 Disasters like those in Boston and Texas led to the establishment of centers for the care of burn victims and to research into the pathophysiology of burn injury.
Inhalation injury can be classified as 1) upper airway injury, 2) lower airway injury, 3) pulmonary parenchyma injury, and 4) systemic toxicity. The extent of inhalation damage depends on the fire environment: the ignition source, temperature, concentration, and solubility of the toxic gases generated. For instance, thermal and chemical compounds usually cause upper airway injury. The water-soluble materials such as acrolein and the other aldehydes damage the proximal airways and set off reactions that are inflammatory to the bronchi and parenchyma, whereas agents with lower water solubility, such as chlorine, phosgene, and nitrogen oxide, nitrogen dioxide or N2O3 or even N2O4, are more likely to cause insidious injury.24
Pathophysiology
Injury to the oropharynx
Much of the pathophysiology that occurs with inhalation injury is related to edema formation in the oropharynx, bronchial areas and parenchyma, and results from an increased transvascular fluid flux from these respective vascular beds. Before a discussion of the changes that occur in these structures following inhalation injury, a review of the forces responsible for the variables of the Starling–Landis equation should be reviewed:25,26
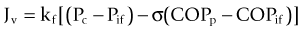
The major pathophysiology seen in the oropharynx following inhalation injury is induced by microvascular changes similar to those seen with thermal injury in other areas of the body. The heat denatures protein, which in turn activates complement. Complement activation causes the release of histamine.27,28 Histamine then causes the formation of xanthine oxidase, an enzyme involved in the breakdown of purines to uric acid.29 During this conversion, reactive oxygen species (ROS) are released.30,31 Reactive oxygen species combine with NO, constitutively formed in the endothelium, to form reactive nitrogen species (RNS).32 The latter produce edema in the burned area by increasing the microvascular pressure and permeability to protein.33,34 Eicosanoids are also released35,36 which, along with oxygen free radicals and IL-8, attract polymorphonuclear cells to the area.37 These neutrophils then amplify the release of oxygen radicals, proteases and other materials into burned areas (Fig. 18.2).
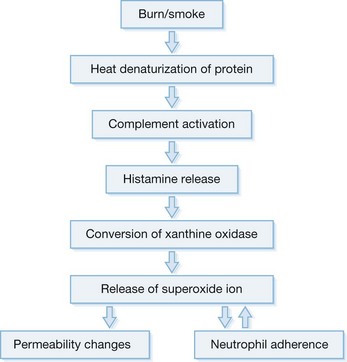
The massive edema occurring in the soft tissue of the oropharynx following burns involves most of the variables in the Starling equation. There is a large increase in microvascular hydrostatic pressure,38 a decrease in interstitial hydrostatic pressure,39 a fall in the reflection coefficient,38 and an increase in interstitial oncotic pressure.39,40 The usual treatment for burn resuscitation calls for the administration of large amounts of crystalloid solutions, which has the effect of reducing the plasma oncotic pressure.41,42 This reduction not only affects the oncotic pressure gradient in the microcirculation but also has been reported to increase the filtration coefficient.43,44 The result of this almost complete breakdown in control of the microvascular function and the insult of fluid administration is massive edema. This is probably nowhere more apparent than in soft tissues of the face and oropharynx. The danger to the patient is extreme. The edema may obstruct the airway, not only making it laborious or impossible to breathe but also making it difficult for the anesthesiologist to intubate the patient (Fig. 18.3A). To avoid this problem, many units prophylactically perform tracheostomies on those patients who have evidence of thermal injury to the upper airway on admission. However, tracheostomy itself may present problems. The tube may further damage injured areas, especially the larynx.45 It may be time to reconsider some of these practices. Perhaps some consideration should be given to fluid resuscitation with colloids, which can prevent some of this soft tissue edema and reduce the volume of fluids required for resuscitation.46,47
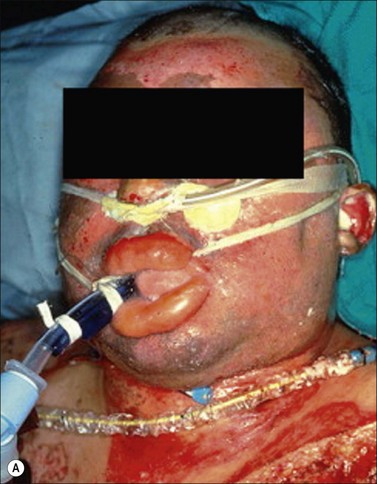
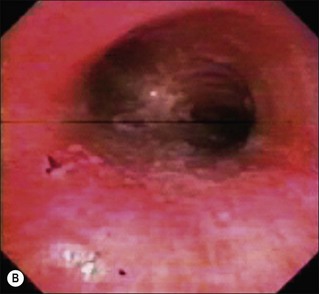
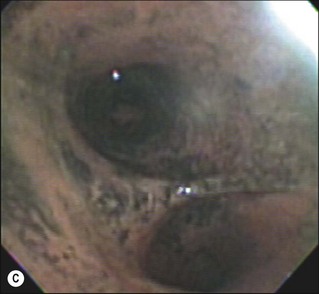
Figure 18.3 Facial and airway injury in the patient after burn and smoke inhalation. (a) A facial burn often associated with thermal injury to the upper airway.153 (b) Hyperemia of airway epithelium. (c) Formation of airway obstructive cast.
(A, from Cancio LC. Airway management and smoke inhalation injury in the burn patient. Clin Plast Surg. 2009; 36(4): 555–67.)
Injury to the tracheobronchial area
With rare exceptions such as inhalation of steam, the injury to the airway is usually due to the chemicals in smoke. The heat capacity of air is low and the bronchial circulation very efficient in warming or cooling the airway gases, so that most gases are at body temperature as they pass the glottis.48 Flames must be in almost direct contact with the airway to induce thermal injury.49 The chemicals in smoke are dependent on the materials that are being burned; however, for the most part the host response is similar. In most instances biological materials such as cotton fabric, wood, grass, or products of these such as cattle feces (commonly used as fuel in Third World countries) are the fuel for the fire. These contain caustic materials such as ROS and RNS, organic acids and aldehydes.50 These chemicals interact with the airway to induce an initial response to trigger an inflammatory response.
Many of the studies that have been reported relative to the bronchial circulation following smoke inhalation injury have been performed in sheep, because these animals have a single bronchial artery51 and have a single lymphatic draining the lung that allows the measure of pulmonary transvascular fluid flux.52 There was a 10-fold increase in bronchial blood flow within 20 minutes of smoke inhalation.53 These same animals demonstrate a sixfold increase in pulmonary transvascular fluid flux and a fall in PaO2/FiO2 ≤ 200, but these were delayed to 24 hours. Similar findings have been reported in patients with smoke inhalation alone or the combination of a large cutaneous thermal injury and smoke inhalation.54
Hyperemia of the airway is such a consistent finding in smoke inhalation that it is used to diagnose the injury.55,56 Other variables that are used include injury in an enclosed space, singed nasal hair and soot in sputum. However, these latter injuries may be present but the subject may still not develop the signs of low Pa and pulmonary edema characteristic of inhalation injury. Airway inflammation plays a major role in the overall response to inhalation injury (Fig. 18.3B and 3C).
As we have noted, there is a large sustained increase in blood flow in the airway following smoke inhalation.57 These changes in blood flow were associated with increased bronchial microvascular permeability to protein and small particles58 and pressure.59 Simultaneously with the changes in the function of the bronchial microvasculature, there is a loss or shedding of the bronchial columnar epithelium.60,61 These changes result in a perfuse transudate with a protein content similar to an ultrafiltrate of the plasma.62 There are also copious secretions from the goblet cells.63 Early in the response these secretions are fluid and form a foamy material in the airway that many have mistaken for severe pulmonary edema in humans.64 After several hours this transudate/exudate solidifies or clots, forming obstructive material in the airways65 (Table 18.1). These obstructive materials formed in the upper airway may appear in the lower airway and alveoli.63 This is problematic in several respects. In some rare instances of severe airway injury these materials can induce total obstruction (Fig. 18.4). Occlusion of some of the bronchi or bronchioles in the setting of high production of NO can lead to a loss of hypoxic pulmonary vasoconstriction and hence increased shunt fraction. Loss of hypoxic pulmonary vasoconstriction with inhalation injury has been reported. Lastly, if single bronchi are occluded while the patient is on a volume-limited ventilator there could be overstretch, and barotrauma to the alveoli of the non-occluded portion of the lung can occur.
Table 18.1 Mean levels of airway obstruction in uninjured sheep and 48h after burn, smoke inhalation, and combined smoke inhalation and burn injury
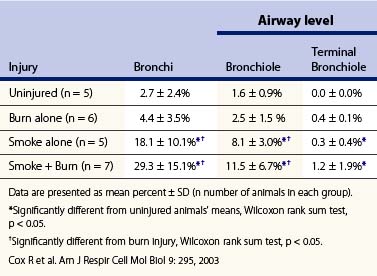
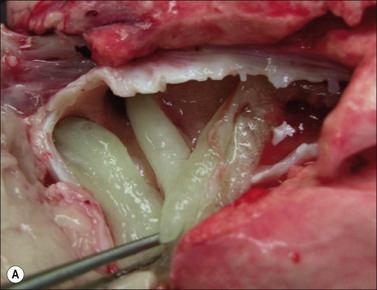
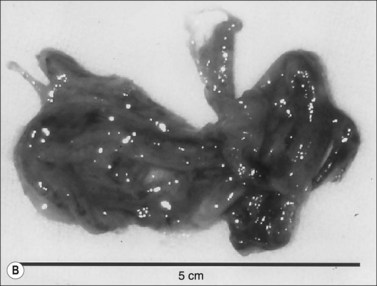
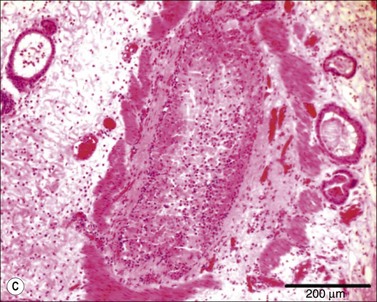
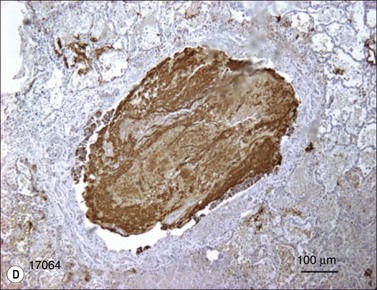
Figure 18.4 Airway obstructive cast. (A) Macroscopic pictures of airway obstructive cast in sheep 48 hours after burn and smoke inhalation injury. (B) Macroscopic picture of airway cast taken from patient with burn and smoke inhalation injury by bronchoscope.154 (C and D) Microscopic pictures of bronchi in sheep and bronchioles in a patient, totally blocked by obstructive cast after burn and smoke inhalation injury.
(A & B from Nakae H, Tanaka H, Inaba H. Failure to clear casts and secretions following inhalation injury can be dangerous: report of a case. Burns. 2001; 27(2): 189–91.)
The airway is richly innervated with vasomotor and sensory nerve endings.66 It is also known that these fibers release neuropeptides that can produce inflammatory responses.67 Neuroinflammation is responsible for pathophysiology in a number of clinical situations, including tissue injury induced by chemicals.68,69 Lange et al. reported that antagonists to substance P and calcitonin gene-related peptide had a marked effect on the response when administered to sheep and mice that were injured with both burn and smoke inhalation.70,71 In the ovine model the combination of burn and smoke inhalation injury caused a 10-fold increase in pulmonary transvascular fluid flux and a reduction of PaO2/FiO2 to ≤200. These changes were reversed by neuropeptide receptors blocking agents.70 Neuropeptide release can cause activation of nitric oxide synthase, have chemokine activity, and alter microvascular permeability.72 The resultant activities lead to the formation of reactive oxygen and nitrogen species.73 Some of the latter are very potent oxidants that can damage DNA.32 Damage to DNA causes the activation of a repair enzyme poly-(ADP-ribose) polymerase (PARP).74 This enzyme depletes the cell of high-energy phosphates and causes the activation of nuclear factor-κB (NF-κB).75,76 Activation of the nuclear factor causes the upregulation of iNOS and IL-8, thereby accelerating the production of reactive oxygen and nitrogen species.77 NO and 3-nitrotyrosine, an index of reactive nitrogen species, iNOS mRNA and protein, have been reported to be in the airway after smoke inhalation.73,78 Compounds that catalyze the breakdown of peroxynitrite reduce the response to smoke inhalation. Poly-(ADP ribose) (PAR), the product of the constitutive enzyme poly-(ADP ribose) polymerase, has been identified in airway tissues following smoke inhalation.73 Inhibition of PARP prevented the formation of PAR, the upregulation of NF-κB and the formation of 3-nitrotyrosine.79 Similarly, Lange et al. have reported that compounds that inhibit peroxynitrite by catalyzing its rapid breakdown likewise prevented the formation of these materials.80 It is interesting to note that mice lacking the PARP genes or given a PARP inhibitor will not show the typical inflammatory changes usually observed with asthma.81 Thus in many ways inhalation of smoke may be similar to forms of airway injury. The fact that the response to inhalation injury is driven by neuroinflammation suggests that the response to smoke from wood or cotton should be similar.
Injury to the lung parenchyma
As noted above, the lung parenchyma changes produced by burn and smoke inhalation, as reflected by reduced PaO2/FiO2 and reduced compliance, and increased edema formation, are delayed.82 This delay is dependent on the severity of the airway injury.50,83 Lung injury is associated with an increased pulmonary transvascular fluid flux.84 The degree of transvascular fluid is proportional to the duration of smoke exposure50 and is not caused by CO in the inhalant gas.85 However, the degree of arterial CO is related to severity of inhalation injury.86 The factors responsible for fluid leak are codified in the Starling–Landis equation.25,26 The variables of this equation relate fluid movement to pressure and permeability variations. With inhalation of smoke there is a reduction in refection coefficient (permeability to protein), an increase in filtration coefficient (permeability to small particles), and an increase in pulmonary microvascular pressure.87,88 Animals that had been exposed to smoke inhalation injury were also noted to have reduced PaO2/FiO2. These variables are more severely affected when the inhalation is combined with burn injury.89 The change in this variable showed a good relationship with the histology injury scores and the changes in transvascular fluid flux.79 In addition, there was a loss of hypoxic pulmonary vasoconstriction in the injured animals that would help to explain the loss of oxygenation.90
As in the oropharynx the injury is associated with activation of PARP and 3-nitrotyrosine and is markedly reduced by the administration of an iNOS or PARP inhibitors.78
The venous outflow of the bronchial circulation drains into the pulmonary microcirculation at the pre-capillary level.91 Considering the fact that initial damage to the airway appeared to drive the pathophysiology of the parenchyma, investigators hypothesized that the bronchial blood might deliver cytotoxic materials or cells into the pulmonary microcirculation. To test this hypothesis, several investigators have tied off the bronchial artery of sheep and then exposed the animals to smoke.58,82,92 In these studies the hypothesis was confirmed and the lung parenchymal changes were reduced.
What could be the linkage between the airway, the bronchial venous drainage and parenchymal injury to the lung? Neutrophils activated in the bronchial circulation flow out into the bronchial venous drainage. Activated polymorphonuclear cells (PMN), especially neutrophils, are stiff. The diameter of neutrophils that have been fixed is approximately 7 µm.93 Because these cells have been dehydrated in alcohol as part of the fixation process, unfixed cells are much larger, of the order of 12 µm. The pulmonary capillary is small, with an average diameter of 6 µm.93 Normally, the large neutrophil can traverse the pulmonary capillary by changing shape. However, many of the neutrophils have been activated in the bronchial areas, their F-actin is activated, and the cells are stiff and cannot deform. These stiff cells are carried to the pulmonary microvasculature where they are impaled by the narrow pulmonary capillaries. The activated neutrophils release reactive oxygen species and proteases that damage the parenchyma. The following evidence supports this concept of neutrophil cytotoxicity. Oxidative processes are well known following inhalation injury. There is lipid peroxidation and release of proteolytic enzymes following injury.94–96 Administration of protease inhibitors or scavengers of reactive oxygen species will reduce the response to smoke inhalation95,97–99 when activated PMNs lose the L-selectin on their surface. This L-selectin shedding is prevented by the treatment with an L-selectin antibody.100 Treatment of the cells with an antibody to L-selectin will prevent the changes in transvascular fluid flux and other aspects of parenchymal damage.101 The final proof of this hypothesis was to deplete the animals of their neutrophils and determine how this affected their response to inhalation injury. In these studies of sheep depleted of their leukocytes, a high percentage of the response to smoke inhalation was blocked.102 The pathophysiology of acute lung injury secondary to burn and smoke inhalation injury is summarized in Figure 18.5.

In addition to the depletion of antioxidants discussed above, it has also been reported that burned patients are depleted of arginine.103 When arginine levels are low the nitric oxide synthase produces superoxide rather than nitric oxide.104 Following smoke inhalation, arginase activity is also elevated.105 This enzyme also depletes arginine by converting it to ornithine.106 The administration of arginine may assist in reducing the oxidation that occurs with inhalation injury.107 However, the necessity of administering the arginine as arginine hydrochloride (because of solubility) limits the amount that may be given intravenously without producing acidosis.
Long-term effects of inhalation injury
When earlier editions of this book were published mortality from inhalation injury was high and the acute inflammatory aspects of the injury were considered a major vector of mortality. Now 85% of patients survive inhalation injury4 and so the long-term aspects of the injury have increased. When our patients are examined years after burn injury they demonstrate restrictive lung disease and reduced diffusion capacity, signs of fibrotic lung disease.108 At autopsy patients and animals both reportedly show hyaline membrane and deposition of collagen in their lungs in a similar fashion to other forms of acute lung injury.105,109 As stated above, two enzymes compete for arginine, nitric oxide synthase and arginase.110 NOS forms NO and RNS. Arginase forms ornithine, which is converted into polyamines and proline, leading to the formation of collagen.111 When NOS is active it forms N-(ω)-hydroxy-nor–l-arginine (NOHA). NOHA breaks down into NO and citrulline. NOHA is a potent inhibitor of arginase.112,113 Thus as long as the activity of NOS is elevated, arginase is inhibited. It has recently been reported that the endogenous inhibitor of NOS, asymmetric dimethylarginine (ADMA), begins to increase in the lung of sheep following combined burn and smoke inhalation injury.105
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


