Fig. 2.1
Schematic structure of the skin and immune cells. Various immune cells reside in the two major compartments of the skin, the dermis and the epidermis. Epidermal dendritic cells (Langerhans cells) represent the main resident antigen-presenting cell in the epidermis; a few CD8+ T cells can be found primarily in the basal layers of the epidermis. The dermis contains different subtypes of dermal dendritic cells (not further specified), CD4+ T cells, macrophages, and mast cells. Migrating immune cells traffic through dermal blood vessels and lymphatic conduits (not shown)
2.3 Autoimmune Bullous Diseases
Autoimmune bullous diseases (AIBD) are a group of rare acquired organ-specific autoimmune diseases, pathogenetically characterized by the presence of autoantibodies directed against adhesion molecules of the skin and mucous membranes [8–10]. These autoantibodies trigger a cascade of events which finally result in either loss of adhesion of epidermal keratinocytes (pemphigus diseases) or in loss of cellular and extracellular integrity at the basement membrane zone (pemphigoids, epidermolysis bullosa acquisita, etc.), resulting in split formation and clinically apparent blisters and painful erosions [11]. AIBD are categorized into several subgroups based on clinical characteristics and histopathological analysis, and they can be classified into further details by their target autoantigens and the circulating autoantibody profile. Recent findings in the field of AIBD research provided notable advances in our understanding of their pathophysiology, including the etiology, genetic risk factors, epidemiology, and immune pathology. These advances were based on clinical studies using techniques such as immunoblotting, immunofluorescence analysis, ELISA, and flow cytometry. Moreover, preclinical models and especially animal model studies revealed basic insight into autoimmune mechanisms leading to loss of self-tolerance to the identified autoantigens in these disorders [12–15]. Recently, severity indices, disease activity scores, and clinical definitions have been introduced for some of the most important AIBD, such as pemphigus and bullous pemphigoid, which provide a mandatory prerequisite for conducting international multicenter trials in these rare autoimmune disorders [16–19].
2.4 Pemphigus Diseases
2.4.1 Pemphigus
Pemphigus diseases include a group of severe AIBD characterized by the loss of epidermal keratinocyte adhesion (acantholysis) resulting in intraepidermal split formation in the skin and mucous membranes. Clinically, pemphigus patients demonstrate flaccid blisters and crusty erosions on the skin and painful erosive defects at the mucous membranes. Pemphigus is considered as a paradigm of an autoantibody-mediated autoimmune disorder, since the passive transfer of patients’ IgG is sufficient to induce a blistering phenotype in neonatal mice (passive transfer model) [20]. Clinically, neonatal pemphigus, i.e., the transient development of blisters in neonates whose mothers suffer from active pemphigus, is explained by the placental transmission of maternal autoantibodies [21]. The desmosomal cadherins desmoglein 1 (Dsg1) and Dsg3 are well-defined target antigens in pemphigus [9]; however, recent studies identified IgG autoantibodies against a variety of non-desmosomal autoantigens, among others acetylcholine receptors, annexins, non-desmosomal adhesion molecules, as well as intracellular targets such as mitochondrial components [22, 23]. The pathogenic relevance of the non-desmosomal autoantibodies remains to be shown in further details [24]. Except for IgA pemphigus, circulating autoantibodies are of the IgG isotype of which IgG4 correlates very well with disease activity [25, 26]. Recently, Dsg3-specific IgE autoantibodies have been detected in acute pemphigus patients [27]. The exact mechanism of how binding of these autoantibodies to their target structures finally leads to loss of keratinocyte adhesion is still unclear [28]. Different hypotheses including steric hindrance of desmosomal Dsg interactions, binding to non-desmosomal Dsg proteins resulting in reduced recruitment of non-desmosomal Dsg to the desmosomes, endocytosis of autoantibody-Dsg complexes, activation of various signaling pathways, and induction of apoptosis are being investigated at the moment [28–33]. Pemphigus can be divided into several subtypes of which pemphigus vulgaris (PV) and pemphigus foliaceus (PF) constitute the major types [34]. Among other very rare variants, paraneoplastic pemphigus (PNP) is characterized by severe mucosal involvement and bronchiolitis obliterans and often associated with underlying lymphoproliferative malignancies. The clinical hallmarks of IgA pemphigus are annular vesicles and pustules, crusty erosions, and erythematous plaques [35].
2.4.2 Pemphigus Vulgaris (PV)
Pemphigus vulgaris (PV) is the most prevalent pemphigus subtype which typically shows mucosal and mucocutaneous involvement, respectively. In PV, Dsg3 is the best characterized major autoantigen, so far [36, 37]. Depending on their clinical phenotype, most PV patients demonstrate circulating IgG1 and IgG4 antibodies against Dsg3 or both Dsg1 and Dsg3 protein [38, 39]. The so-called desmoglein compensation theory aims at correlating the clinical phenotype (mucosal, mucocutaneous) with the autoantibody profile, i.e., whether only anti-Dsg3-IgG or in combination with anti-Dsg1-IgG is present [34]. In contrast to subepidermal blistering autoimmune dermatoses, such as bullous pemphigoid, in PV, histopathology of lesional skin shows a sparse infiltration of inflammatory cells; in early skin lesions, an eosinophilic infiltrate might be seen (eosinophilic spongiosis). Considering the pathogenic relevance of IgG autoantibodies in PV, autoreactive CD4+ T cells and B cells are considered as crucial immune cells in this disease [40–44]. Although direct experimental evidence is missing, it seems likely that the activation of autoreactive CD4+ T and B cells takes place in skin draining lymph nodes. Autoreactive, Dsg3-specific CD4+ T cells have been isolated and functionally characterized from peripheral blood of PV patients [41, 45–48]. The Dsg3-specific activation of these autoreactive T cell clones is dependent on the HLA-class II haplotype that is highly prevalent in PV patients, i.e., HLA-DRB1*0402, HLA-DQB1*0503 [49, 50]. Therapeutic depletion of B-cells using the anti-CD20 monoclonal antibody (rituximab) leads to a dramatic decline of circulating anti-Dsg3-IgG followed by a significant clinical improvement in PV patients [43, 51–54], suggesting that autoantibody-secreting plasma cells in PV are rather short-lived compared to plasma cells releasing pathogen-reactive IgG [55, 56].
2.4.3 Pemphigus Foliaceus (PF)
Pemphigus foliaceus (PF) is histologically characterized by the loss of keratinocyte adhesion in the subcorneal layer of the epidermis, resulting in superficial blister formation. Early skin lesions might reveal eosinophilic and neutrophilic spongiosis [57]. PF patients present circulating autoantibodies, mainly of the IgG1 and IgG4 subclasses, against the desmosomal glycoprotein desmoglein 1 (Dsg1) [9, 25]. Anti-Dsg1-IgG4 has been shown to be associated with active PF disease [58]. Clinically, superficial blisters and scaly, crusted erosions develop in the seborrheic areas, i.e., face, scalp, and trunk, while the mucous membranes are typically not involved [59, 60]. Dsg1-reactive CD4+ T cell responses in peripheral blood of patients with the endemic form of PF, fogo selvagem, have been identified [61]. In this study, memory CD4+ Th2 cells responded to the extracellular domain of the Dsg1 protein in an HLA-DR-restricted manner [61]. Gebhardt et al. showed that both PF patients and HLA-DR/–DQ-matched healthy individuals exhibit Th1 and Th2 responses to the recombinant ectodomain of Dsg1 [62], suggesting that the presence of CD4+ autoreactive T cells in peripheral blood is not restricted to PF and PV patients, respectively [48, 62]. Fogo selvagem (FS) is an endemic form of pemphigus foliaceus (PF), in which patients develop mainly IgG4 autoantibodies specific for Dsg1 [63, 64]. Moreover, in acute disease, FS patients demonstrate autoantibodies directed against NH2-terminal epitopes in the extracellular domain 1 (EC1) and EC2, whereas patients at preclinical stages predominantly show reactivity with COOH-terminal epitopes [65]. Recently, Qian et al. provided evidence that salivary gland antigens of the sandfly, specifically the LJM11 salivary protein, is recognized by anti-Dsg1-IgG of FS patients [66]. The cross-reactivity with LJM11 salivary protein in genetically susceptible individuals might represent an environmental trigger to initiate this autoimmune disease [66].
2.4.4 Pemphigus Erythematosus
Originally, pemphigus erythematosus (PE) (also known as Senear-Usher syndrome) was described as a clinical subtype of PF presenting characteristics of lupus erythematosus [67]. Although several cases of PE have been described, the criteria for making the diagnosis of PE are controversial and not well defined yet. PE patients present with erosions on the seborrheic areas and erythema in the malar region. Immunoserologically, PE patients demonstrate circulating IgG autoantibodies that are reactive with Dsg1, as in PF [68]. However, the autoantibody profile in PE seems to be more diverse, since additional autoantibodies reactive with components of the hemidesmosome, such as the BP230 antigen and plakins, have been described [69].
2.4.5 Paraneoplastic Pemphigus
This rare pemphigus subtype is considered as an autoimmune multiorgan syndrome associated with an underlying neoplasia, mostly hematological disorders such as non-Hodgkin lymphoma, chronic lymphocytic leukemia, Castleman disease, or non-hematological malignancies, such as adenocarcinomas [70, 71]. The cutaneous involvement is variable including erosions, widespread blisters, erythema multiforme-like lesions, and lichenoid manifestation, whereas mucous membranes demonstrate severe and extensive mucositis and hemorrhagic erosions and crusts [72]. The autoantibody response in paraneoplastic pemphigus is more diverse compared to PV and PF, including IgG against desmoplakin, envoplakin, periplakin, plectin, and the recently identified 170-kDa antigen, the protease inhibitor alpha 2 macroglobulin-like-1 [73]. In contrast to the abovementioned pemphigus subtypes, in paraneoplastic pemphigus, histopathological findings might be as diverse as the clinical manifestation. Intraepidermal suprabasilar split formation is usually present as well as mononuclear infiltrates and intense interface dermatitis with vacuolar degeneration of the basal keratinocytes, respectively [71].
2.4.6 IgA Pemphigus
Immunoglobulin A (IgA) pemphigus is characterized by tissue-bound IgA autoantibody deposits on the cell surface of epidermal keratinocytes [74, 75]. Depending on clinical manifestations, histopathological findings, and immunofluorescence staining patterns, the two subtypes of IgA pemphigus, subcorneal pustular dermatosis (SPD) and intraepithelial neutrophilic IgA dermatosis (IEN), can be distinguished [75]. Annular erythematous plaques, erythema with peripheral subcorneal pustules predominantly in the intertrigines (SPD) and erythematous papules, vesicles, erosions, and crusts (IEN) represent typical clinical findings in IgA pemphigus patients. Circulating IgA autoantibodies that are reactive with desmosomal cadherins have been identified in these patients [74]. In SPD-type IgA pemphigus, autoantibodies targeting the desmosomal proteins Dsc 1, 2, and 3 have been described [76, 77]. In IEN-type IgA pemphigus, autoantibodies directed against the classical autoantigens in pemphigus, i.e., Dsg1 and Dsg3, have also been identified [74, 78–80]. However, there is no distinct autoantibody profile depending on the subtype of IgA pemphigus. Recently, a case of SPD-type IgA pemphigus demonstrating IgA autoantibodies against Dsc2 and Dsc3 as well as BP180-reactive IgA autoantibodies, suggestive of linear IgA dermatosis (LAD), has been reported [81]. The prevalence of IgA autoantibodies in contrast to the major subtypes of pemphigus, PV and PF, which are characterized by tissue-bound and circulating IgG autoantibodies, remains unclear in IgA blistering diseases.
2.4.7 Pemphigus Herpetiformis
Pemphigus herpetiformis (PH) is another rare but distinct variant of pemphigus with clinical characteristics resembling dermatitis herpetiformis, such as pruritic annular erythemas with peripheral vesicles and annular-shaped papular lesions [11, 82, 83]. In most PH patients, circulating anti-Dsg1-IgG autoantibodies and in fewer cases anti-Dsg3-IgG have been detected [84]. Recently, patients with PH-like, atypical pemphigus who developed autoantibodies against non-desmoglein desmosomal targets, such as Dsc1 [85] and Dsc3, have been described as well [86, 87]. In a large immunoserological study, Ohyama et al. (2012) showed that sera of 15 PH patients reacted predominantly with Dsg1- and Dsg3-epitopes located in the NH2-terminal regions of these autoantigens [88]. So far there is no sound explanation for the “atypical clinical manifestation” and the autoantibody profile in PH compared with the classical pemphigus subtypes.
2.5 Subepidermal Bullous Diseases
2.5.1 Bullous Pemphigoid
Bullous pemphigoid (BP) is the most common autoimmune blistering skin disease in adults with an increasing incidence in elderly patients; incidences in European countries of 12–22 per one million people per year have been reported [89–91]. BP is characterized by the presence of IgG autoantibodies specific for the hemidesmosomal antigens BP230 and BP180 (collagen XVII) (Fig. 2.2) [92–94]. In a multicenter prospective study, Di Zenzo and colleagues [95] investigated the autoantibody reactivity to these two major autoantigens in a cohort of 35 BP patients [95]. In this study, the authors demonstrated that during the course of the disease, both intramolecular and intermolecular epitope spreading events occurred in BP. Moreover, the autoantibody reactivity correlated with disease activity and severity in these patients [95]. In addition, autoreactive T lymphocytes recognizing defined epitopes of the BP180 autoantigen have been detected in peripheral blood of BP patients [96]. In this study, T cell and autoantibody reactivities especially to NH2-terminal epitopes of the BP180 protein were related to more extensive disease activity [96]. In addition to BP180-/BP230-specific IgG autoantibodies, the pathogenic relevance of BP180-reactive IgE autoantibodies has been well characterized in BP, suggesting that T helper 2 cells (Th2) play an important role in activating autoreactive B cell clones to secrete BP180-reactive autoantibodies [97–100]. Clinically, the early (pre-bullous) stage of BP is dominated by urticarial papules and plaques showing edema and eosinophilic infiltration by histopathology [101], suggesting that autoantibodies binding to BP180-expressing basal keratinocytes initiate the autoimmune cascade finally leading to loss of keratinocyte adhesion at the basement membrane zone by activation of complement factors, recruitment of eosinophils and neutrophils, mast-cell degranulation, and the release of various proteases. In experimental animal models of BP, passive transfer of antibodies recognizing the non-collagenous domain 16A (NC16A) of BP180 induced a bullous pemphigoid-like phenotype depending on complement activation, neutrophil infiltration, and mast cell degranulation [102–104].
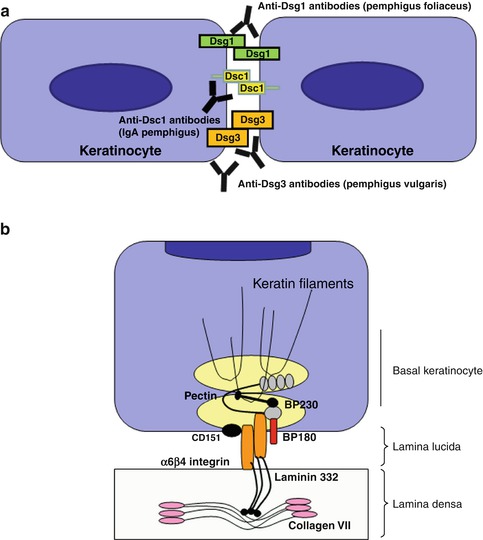
Fig. 2.2
Schematic illustration of major target antigens in autoimmune blistering diseases. In autoimmune blistering diseases, autoantibodies are directed against adhesion molecules conferring desmosomal cell-cell adhesion (a) and cell-matrix interactions at the basement membrane (b). (a) The desmosomal cadherins are composed of desmogleins and desmocollins; adhesion of neighboring epidermal keratinocytes is based on homo- and heterophilic interaction of the extracellular domains. Autoantibodies of representative pemphigus diseases are illustrated, such as anti-desmoglein 3 antibodies in pemphigus vulgaris and anti-desmoglein 1 IgG in pemphigus foliaceus. Desmocollins are identified as autoantigens in rare “atypical variants” of pemphigus, such as IgA pemphigus as shown for anti-desmocollin 1 IgA autoantibodies. (b) Basal keratinocytes are attached to the basement membrane by hemidesmosomes. The intracellular plaque protein BP230 is targeted among others in bullous pemphigoid; BP180 and α[alpha]6ß[beta]4 integrin bind to the extracellular matrix protein laminin 332, a major component of the lamina densa. These are important autoantigens in bullous pemphigoid (BP180) and mucous membrane pemphigoid (α[alpha]6ß[beta]4 integrin, laminin 332). Type VII collagen is an anchoring fibril that connects the lamina densa to the underlying collagen fibers of the dermis; it is targeted by autoantibodies in epidermolysis bullosa acquisita. The tetraspanin CD151 forms stable laminin-binding complexes with α[alpha]6β[beta]4 integrins in the lamina lucida
2.5.2 Linear IgA Disease
The characteristic autoimmune finding in linear IgA disease (LAD) is the linear deposition of IgA at the basement membrane zone. It is the most common subepidermal blistering disorder in children, and it rarely occurs in adults. Clinically, patients present with urticarial plaques, tense blisters, and erosions often in an annular-shaped pattern (“string of pearls sign”), and more than two thirds of the patients exhibit mucosal involvement, mostly erosions of the oral cavity [105]. The majority of LAD sera recognize a 97 kDa extracellular portion of BP180 in skin extracts [106] and the 120 kDa shed ectodomain (LAD-1 antigen) of BP180 [107]. With regard to the autoantibody profile, i.e., the detection of both IgA and IgG autoantibodies in LAD patients, and the clinical presentation, there seem to be overlapping pemphigoid diseases.
2.5.3 Pemphigoid Gestationis
Pemphigoid gestationis (PG) is a rare pregnancy-associated autoimmune bullous disease that typically appears in the second to third trimester and rarely after birth. Most patients develop autoantibodies against the major BP epitope, the NC16A domain of BP180, and less frequently against BP230 [108, 109]. Autoantibodies of the complement-fixing IgG1 and IgG3 immunoglobulin subclasses are primarily found in PG patients [110]. The autoantigen BP180 is expressed in the amniotic membrane; although the pathophysiology of PG has not been completely clarified, there is evidence that the aberrant expression of disease-associated HLA-DR molecules in the placenta might lead to increased presentation of self-antigens, such as the ectodomain of BP180, followed by the activation of BP180-reactive T and B cells finally resulting the production of BP180-specific IgG. Clinically, PG patients suffer initially from pruritic erythematous urticarial papules and plaques typically located on the abdomen periumbilically and later involving the whole abdomen and thighs. During the course of the disease, clustered tense blisters might develop [111].
2.5.4 Mucous Membrane Pemphigoid
Mucous membrane pemphigoid (MMP) is defined as a mucous membrane-dominated, subepidermal chronic autoimmune blistering disease with autoantibodies directed against the dermal-epidermal junction [112]. MMP mostly affects the oral cavity, conjunctivae, nasal cavity, pharynx, and the anogenital region, and about 30 % of the patients demonstrate cutaneous involvement [113]. Autoantibodies from MMP patients usually react with the C-terminal domain of BP180; other less frequent target antigens include laminin 332 (formerly laminin 5, epiligrin), BP230, alpha6beta4 integrin, and type VII collagen [11, 114, 115]. There is an increased risk in MMP patients with laminin-332-reactive autoantibodies for developing malignancies [116]. Scarring is a characteristic clinical finding in MMP which might lead to blindness in ocular pemphigoid. In more than half of MMP patients, IgA autoantibodies can be detected as well [117, 118]; interestingly the presence of both IgA and IgG autoantibodies has been correlated with a more severe disease activity compared with IgG autoantibodies only [119]. In subsets of MMP patients, the clinical manifestation has been associated with distinct autoantigen reactivity, such as autoantibodies to alpha 6 integrin are common in patients with oral mucosa involvement and reactivity to beta 4 integrin is often seen in ocular pemphigoid [120].
2.5.5 Anti-p200/Anti-lamininγ[Gamma]1 Pemphigoid
Anti-p200 pemphigoid is a very rare subepidermal blistering autoimmune disorder which closely resembles BP and the inflammatory subtype of EBA, respectively. Psoriasis has been described in about a third of Japanese patients diagnosed with anti-p200 pemphigoid [121]. Compared with BP, anti-p200 pemphigoid occurs at younger age, and mucous membrane involvement has been reported in about 20 % of the cases [122]. Indirect immunofluorescence microscopy using human saline-split-skin sections reveals IgG autoantibody binding to the dermal site of the artificial split, thus differentiating anti-p200 pemphigoid from BP. Circulating autoantibodies in these patients recognize a 200-kD protein of human dermal extracts. Recent immunoserological studies demonstrated that about 90 % of anti-p200 pemphigoid sera bind to the COOH-terminus of the laminin γ[gamma]1 chain by Western blotting [123]. Laminin γ[gamma]1 belongs to a family of extracellular matrix glycoproteins that are non-collagenous components of basement membranes that interact via nidogen or integrins [124]. The recombinant laminin γ[gamma]1 protein is applied in ELISA and immunoblot analysis for immunoserological diagnostics [125]. The pathogenic relevance of anti-laminin γ[gamma]1 autoantibodies in this disease remains elusive, since patients’ sera that have been depleted of anti-laminin γ[gamma]1 IgG are still able to induce subepidermal split formation in an ex vivo model [126].
2.5.6 Epidermolysis Bullosa Acquisita
Epidermolysis bullosa acquisita (EBA) is a severe subepidermal blistering disease which is characterized by the presence of mostly IgG autoantibodies especially of the IgG1 and IgG4 isotypes [127] targeting the NH2-terminal non-collagenous (NC1) domain of type VII collagen, a major component of the anchoring fibrils at the dermal-epidermal junction [128, 129] (Fig. 2.2). Numerous reports suggest the presence of IgG and IgA autoantibodies against type VII collagen in EBA, and a minority of the patients exhibit IgA autoantibodies, only [130–132]. Depending on the clinics, two subtypes, the classical mechanobullous and the inflammatory variant of EBA, are distinguished. The latter one resembles other pemphigoid diseases, such as BP, linear IgA disease, or MMP, whereas the mechanobullous variant of EBA presents with tense blisters, skin fragility with slight cutaneous inflammation preferentially localized at mechanically prone areas, scarring, and milia formation. Involvement of the mucous membranes is reported in about half of the EBA patients. Studies in a preclinical animal model of EBA and in vitro studies using peripheral blood mononuclear cells (PBMC) of EBA patients showed that autoreactive T and B cells are mandatory for the production of autoantibodies against type VII collagen [133, 134]. Using recombinant human type VII NC1-protein, Müller and colleagues demonstrated that both autoreactive T cells and circulating IgG autoantibodies recognize similar regions of the type VII NC1 domain [134]. Data generated in a well-characterized mouse model of the inflammatory EBA subtype suggests that upon binding of type VII collagen-specific IgG autoantibodies, complement factors (C5a) are activated leading to an influx of neutrophils which are being activated by binding to the Fc-portion of the tissue-bound autoantibodies, and finally activated neutrophils release oxidases and metalloproteases that mediate extracellular protein proteolysis and interfere with anchoring fibrils.
References
1.
Bos JD, Kapsenberg ML. The skin immune system: progress in cutaneous biology. Immunol Today. 1993;14(2):75–8.PubMed
2.
Streilein JW. Skin-associated lymphoid tissues (SALT): origins and functions. J Invest Dermatol. 1983;80(Suppl):12s–6.PubMed
3.
Egawa G, Kabashima K. Skin as a peripheral lymphoid organ: revisiting the concept of skin-associated lymphoid tissues. J Invest Dermatol. 2011;131(11):2178–85.PubMed
4.
Nestle FO, Di Meglio P, Qin JZ, Nickoloff BJ. Skin immune sentinels in health and disease. Nat Rev Immunol. 2009;9(10):679–91.PubMed
5.
Schakel K, Hansel A. News from dendritic cells in atopic dermatitis. Curr Opin Allergy Clin Immunol. 2011;11(5):445–50.PubMed
6.
Di Meglio P, Perera GK, Nestle FO. The multitasking organ: recent insights into skin immune function. Immunity. 2011;35(6):857–69.PubMed
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


