Chapter 25 The hepatic response to thermal injury
 Access the complete reference list online at http://www.expertconsult.com
Access the complete reference list online at http://www.expertconsult.com
Introduction
More than 2 million Americans each year experience a thermal injury – one of the most severe examples of a traumatic injury.1 Worldwide, the World Health Organization (WHO) attributes >330 000 deaths each year to thermal injuries and their sequelae.2 In the USA alone, more than 440 000 children are treated for burn injuries annually.3 Severe burn injuries – the third most common cause of death for American children – account for up to 1100 pediatric deaths each year,4,5 accounting for a significant number of hospital admissions.6,7 The effects induced by these injuries are not limited to the injured area alone. A severe burn injury has a devastating effect on the injured patient by affecting almost every organ system, resulting in heightened morbidity and mortality.8 Amplified availability of glucose leads to increased protein catabolism and lipolysis, initiating the post-burn hypermetabolic stress response.7–9 Systemic inflammation, including pathophysiologic regulation of cytokines, hormones, and acute phase proteins, drives the hypermetabolic stress response.10–12 Prolongation or amplification of the hypermetabolic, inflammatory, or acute phase responses may result in dysregulation of counter-regulatory stress hormones (catecholamines, cortisol, glucagon), thereby exacerbating post-burn hypercatabolism, multi-organ failure, and death.11–13
For more than 20 years, focused research efforts on improving post-burn resuscitation, hypermetabolism, infection, ventilation, resuscitation, and wound healing have led to reduced morbidity and mortality.7 Further advances in clinical care, however, are needed to reduce morbidity and mortality even further. Through a series of studies, we have built a foundation that supports the hypothesis that the liver is a central player in the response to burn.14–17 Through the modulation of immune, inflammatory, metabolic, and acute phase response signal transduction pathways, the liver contributes greatly to survival and recovery following a severe burn injury.13 Because of the dearth of information delineating the liver’s role after a thermal injury, we have spent the past 15 years engaging in studies to shed light on this topic. This chapter discusses liver function under normal conditions and following a severe insult such as a burn injury.
Anatomy, function, and physiology of the liver
Anatomy
In an average-sized adult, the liver weighs approximately 1500 g, making up almost 2% of total body weight. When the liver is injured, such as following a severe burn injury, its size can increase as needed to meet the additional demands. The lobar anatomy is used to divide the liver in the American system, using the intrahepatic location of the branches of the hepatic artery, portal vein and bile ducts. In this method, the left lobe is divided into medial and lateral sections, while the right lobe is divided into anterior and posterior sections. Soupault and Couinaud’s segmental system is used by the French to divide the liver into VIII segments. The hepatic artery and the portal vein are the two sources of the liver’s blood supply. One-quarter of the hepatic blood flow occurs via the hepatic artery, which provides oxygenated blood. Three-quarters of the blood flow through the liver is accounted for by the portal vein, which drains the splanchnic circulation.18
Physiology
A broad spectrum of biological functions is orchestrated by the liver. The fine interrelated physiologic-anatomic units of the liver direct these necessary processes: regulation of energy availability; nutrient management (including storage, distribution, and disposal); substrate synthesis, transformation, and metabolism; and metabolism and removal or toxins and pollutants.18,19
1 The circulatory system. In order to build the pool of metabolites for energy and other functions, the liver extracts nourishment from the blood supply that extracts absorbed nutrients from the intestinal tract.
2 The biliary system. Bilirubin, cholesterol, detoxified drugs, and other liver-secreted substances are eliminated through the biliary system.
3 The mononuclear phagocyte system (MPS) (formerly known as the reticuloendothelial system, RES). A part of the immune system that detects antigens and disposes of senescent cells. Within the liver, Kupffer, endothelial, and phagocytic cells make up 60% of the MPS.
4 The hepatocytes. The metabolic demands of the entire body are met by the body. These cells synthesize, secrete, and store metabolites, and perform catabolic and anabolic functions. The generation of energy by converting adenosine triphosphate (ATP) to adenosine diphosphate (ADP) fuels local and systemic functions.
5 The liver as the source of hormones. The liver is the site of the synthesis and secretion of a variety of hormones including insulin-like growth factor-I (IGF-I), IGF binding proteins (IGFBPs), and hepatocyte growth factor (HGF). In addition to synthesizing and secreting necessary hormones, the liver acts as both a paracrine and an endocrine tissue, thereby amplifying the hepatic role in the hormonal axes.
Circulatory system
The majority of metabolic processes are regulated in one way or another by the liver. To fuel these functions, the liver expends 20% of the body’s total energy supply while consuming up to 25% of total utilized oxygen. These utilizations are not as high as one might expect due to an unfettered blood supply and the unique architecture within the liver. Estimations of the mean total hepatic blood flow range from 100 to 130 mL/kg per min. The majority (three-quarters) of the total hepatic blood flow is via the portal vein, while the remaining quarter is from the hepatic artery. When portal flow is reduced, a reciprocal increase in hepatic arterial blood flow follows; however, the reverse does not occur.13,18 Plasma membrane-bound hepatocellular organelles enable specific functions while concurrently interacting with the extracellular matrix, facilitating exchange of metabolites between the blood compartment and the hepatocytes.18,19 At the same time, the liver manufactures proteins, substrates, enzymes, and other substances for local use as well as for use by distal organs and tissues. Metabolic signals direct the liver to produce substrates needed to meet the energy requirements in other tissues. For example, the liver produces acetoacetate for utilization by brain, muscle, and the kidneys; however, this substrate is not used by the liver. Agonists, hormones, and substrates regulate the hepatic energy-related functions. As the liver receives blood via the arterial and portal circulation, nutrients are processed while toxins are metabolized, and then these are stored for later use, transformed for immediate use, or distributed to their final destination via lymphatic, vascular, or biliary circulations.
Portal venous flow into the liver is largely regulated by extrahepatic factors, including the flow rate from the intestines and spleen, food, bile salts, secretin, cholecystokinin, pentagastrin, epinephrine, vasoactive intestinal peptide, and glucagon. With blood making up 25–30% of the liver volume, the liver acts as a reservoir that can be tapped in time of physiologic need. More than 300 mL can be released into systemic circulation without adversely affecting liver function during acute blood loss. The liver can also store great amounts of blood – up to 1000 mL of blood can be taken up during right-sided heart failure without adversely affecting liver function.18–20
Biliary system
Although bile secretion occurs independently of liver blood flow, this is not the case when a patient is in shock. Bile is either formed at the hepatocyte’s canalicular membrane or by the bile ductules and ducts. Hepatocytes secrete the majority (four-fifths or approximately 1500 mL) of the total daily production of bile, while the bile duct epithelial cells secrete the remaining fifth. Phospholipids, proteins, conjugated bile acids, and cholesterol are the principal organic compounds in bile. Bile is modified by epithelial cells as it passes through the biliary ductules or ducts. Cholangiocytes, the highest cells lining the biliary ductules, seem to be hybrids of hepatocytes and ductular cells due to common functions and architecture. Bile secretion is stimulated by secretin, and the bile is secreted into the gallbladder where it is concentrated and stored under fasting conditions. Concentration of bile within the gallbladder is stimulated principally by cholecystokinin, with absorption of up to 90% of the water occurring within a 4-hour period. Bile is forced into the intestines following gallbladder contraction induced by cholinergic stimulation and subsequent relaxation of the sphincter of Oddi. The majority of the bile salt is absorbed into the enterohepatic circulation. Bile acids are then extracted by the liver and transported back to the canalicular membrane for re-secretion into the biliary system. Complete circulation of the total bile pool occurs 6–10 times/day in addition to 2–3 times per meal. Of the total 2–5 g pool, each day 0.2–0.6 g are lost in the stool; however, this is rapidly replaced by the synthesis of new bile acids.20
When heme breaks down, bilirubin is produced and eliminated in the bile. Accumulation of bilirubin in the tissue or blood may occur with extrahepatic biliary obstruction or hepatocellular disease. The conjugation of bilirubin to albumin protects tissue from bilirubin’s toxicity until it is removed from the circulation by the liver via a carrier transport system. Bilirubin and glucuronide are conjugated within the hepatocyte, and then secreted into the bile. When this complex binds covalently with albumin, delta bilirubin is formed. Bacterial reduction of bilirubin in the intestine yields mesobilirubin and stercobilirubin, together known as urobilirubin, which are then excreted in the stool. Oxidation of urobilinogen to urobilin gives the stool its normal brown color.18–21
Metabolic system
Acute phase response
In order to limit tissue damage and to initiate repair processes, the acute phase response (APR) is triggered following activating events such as a severe burn or trauma.13,22 Pro-inflammatory cytokines are released locally by fibroblasts, endothelial cells, and activated phagocytic cells, which then leads to systemic dispersion of the APR.13,22 The systemic phase of the APR induces a body-wide response encompassing the participation of many organs. The hypothalamus acts to raise body temperature, leading to fever. Steroid hormones are released by the pituitary–adrenal axis. Acute phase proteins are synthesized and secreted by the liver. Further promulgation of the hemopoietic responses is initiated in the bone marrow. Finally, the MPS involves the immune system in the APR, as the lymphocytes are activated to detect foreign antigens.13,22 The interaction between the injury site and the liver, however, is a critical step in the initiation of the APR, as the liver is the primary site of acute phase protein production and modulation of the systemic inflammatory response. The APR is characterized by increased production of positive acute phase proteins such as C-reactive protein, α2-macroglobulin, and haptoglobin, with decreased expression of negative acute phase proteins, including albumin, transthyretin, transferrin, and retinol-binding protein.
Carbohydrate metabolism
Another major function of the liver is to provide a readily available source of energy – glucose – to the adrenal medulla, red blood cells, and the central nervous system. Postprandial intestinal carbohydrate digestion delivers 80% glucose and 20% galactose and fructose to the liver, where galactose and fructose are rapidly converted into glucose. Hepatocytes absorb the glucose and convert it directly into glycogen for storage. Up to 65 g of glycogen per kg of liver mass can be stored in this manner, while excess glucose is converted to fat. Although skeletal muscle also produces glycogen, it is only for use locally by this tissue. The glycogen produced by the liver is unique, in that it is available for use by other tissues when needed. Under fasting or starvation conditions, glycogen conversion is the primary source of glucose for the first 48 hours. After that, liver glycogen stores have been depleted, and proteins and fat are instead mobilized to meet the metabolic demands. The main substrate mobilized in the muscle is alanine, which then is converted into glucose by the liver.23,24
Adequate glucose synthesis is ensured by hepatic glycogenolysis, glycogenesis, and the conversion of galactose into glucose. As a result, hypoglycemia is a rarity and is usually only apparent when coupled with extensive hepatic disease. Hyperglycemia, however, results from deficiencies in glycogenesis and commonly occurs alongside severe liver disease. Under ordinary conditions, lactate is produced in the muscles as a result of anaerobic glycolysis. The lactate is then shuttled to the liver where it is converted back into glucose, which is then sent back to the peripheral tissues. The constant shuttling of lactate and glucose is known as the Cori cycle. The brain must be continually supplied with glucose as it does not participate in the Cori cycle. Under starvation conditions, glucose is supplied to the brain at the expense of muscle proteins. Glucose metabolism is often deranged in liver disease. Patients with cirrhosis of the liver frequently experience a reduction in the portal-systemic shunting, decreasing the hepatocyte exposure to portal blood. The clinical presentation of this occurrence is an abnormal result following an oral glucose tolerance test. As the severity of the liver disease progresses, so does the extent of the dysfunction. In patients with fulminating hepatic failure, massive loss of hepatic mass and hepatocyte function occurs, leading to hypoglycemia as gluconeogenesis ceases.21
Lipid metabolism
Absorption of fat from the gut, lipolysis-induced liberation of fat from adipocytes, and synthesis of fatty acids from amino acids and carbohydrates are the three main sources of free fatty acids (FFA) used by the liver. Triglycerides (TG) are then formed following the etherification of FFA with glycerol. TG export is a direct function of the availability of very low density lipoproteins (VLDL). Lipids accumulate in the liver in the presence of excess FFA; this results from an imbalance in the TG to VLDL ratio and is commonly a sequela of pregnancy, obesity, diabetes, corticosteroid use, or total parenteral nutrition. When inadequate amounts of protein are ingested or absorbed, the liver undergoes fatty alterations as the TG accumulate due to limited protein availability for lipoprotein synthesis.21,25
In addition to TG and FFA, cholesterol and phospholipid synthesis occurs in the liver, and lipid metabolism can be indirectly determined by measuring cholesterol production. The liver is the primary site of cholesterol synthesis, esterification, and excretion. There is a decrease in total cholesterol as well as in the percentage of esterified cholesterol when parenchymal damage is present. Elevations in cholesterol can result from biliary obstruction, biliary cirrhosis, or as a result of toxic reactions to phenothiazine drugs.20,21
Protein metabolism
The liver synthesizes and secretes 17 of the major plasma proteins in addition to the majority of urea produced in the body. Measurement of serum proteins produced by the liver can serve as an index of hepatic function. The liver is the sole producer of serum albumin and α-globulin. The most abundant serum protein is albumin, and its creation accounts for approximately 15% of total hepatic protein synthesis. As a result, serum albumin levels can be used as a marker of liver function, liver disease, or side-effects following therapeutic administration. However, albumin has a half-life of 21 days; therefore, albumin can only be used to monitor reductions in hepatic protein synthesis if the condition has continued for at least 3 weeks. Furthermore, one has to be cautious to interpret albumin levels, as albumin synthesis can be affected by nutrition, hormones, and inflammatory mediators.16,26
In addition to albumin, hepatic cells also produce fibrinogen, prothrombin, and other blood clotting factors. Despite the numerous proteins being produced by the liver, total protein cannot be used to reflect the extent or severity of liver disease. Although albumin concentrations accurately correlate with liver disease, only hepatic cells produce albumin, so decreases in albumin production trigger a compensatory increase in globin concentrations.21
Vitamin metabolism
Vitamin uptake, storage, and mobilization are additional important functions of the liver. The absorption of fat-soluble vitamins (A,E,D, and K) is dependent on bile salts. At 2–6 hours following oral administration, these vitamins can be found in the thoracic duct. Because Vitamin A is stored exclusively in the liver, excess ingestion may be associated with significant hepatic injury. Ito cells (fat-storing cells) are thought to play a role in Vitamin A storage as well. The initial Vitamin D activation step, conversion of Vitamin D3 to 25-hydroxycholecalciferol, occurs in the liver. Coagulation factors II, VII, IX, and X are dependent on Vitamin K, which is essential for the γ-carboxylation, and activation, of these factors (see below).20,21 Vitamin E has recently garnered much attention due to its potent antioxidative properties. Following a severe thermal or traumatic injury, Vitamin E might reduce oxidative stress and subsequent damage.
Coagulation
Many of the blood coagulation factors are produced in the liver. When liver disease is present, these factors may be altered or rendered defective. There is a decrease in Vitamin K resorption and a subsequent reduction in prothrombin synthesis in the jaundiced liver. When the liver is severely damaged, the hepatocellular dysfunction is so extensive that prothrombin is not produced at all. Clinical measurements of prothrombin and prothrombin time can be used to determine whether the synthesis of prothrombin has been impaired. Hepatic disease is also associated with reductions in factors V, VII, IX, and fibrinogen.20,21
Hormonal system
The liver is the site of hormonal synthesis, secretion, or interaction. Production and secretion into the bloodstream of angiotensinogen occurs within the liver. As noted above, Vitamin D is synthesized when cholecalciferol is hydroxylated by the enzyme 25-hydroxylase to become 25-hydroxycholecalciferol. Growth factors that are important for growth and development such as insulin-like growth factor-I (IGF-I) and the IGF binding proteins (IGFBPs) are made and secreted by the liver. Growth hormone (GH), the primary stimulus for the production of IGF-I, is produced by the pituitary gland but acts mainly on the hepatic cells (GH). Finally, synthesis of hepatocyte growth factor (HGF), a major hepatic regenerative growth factor, occurs in the liver.27
The hepatic response to a severe thermal injury
Liver damage and morphological changes
Burn-induced liver injury is variable, and is typically proportional to the severity of the burn injury. Hepatomegaly, or a fatty liver, is a common post-burn finding (Fig. 25.1). These changes can be reversed; however, the significance of these alterations depends on the cause and the extent of the fat accumulation.28 Autopsies of deceased pediatric burn victims have revealed that fatty infiltration of the liver is associated with increased bacterial translocation, liver failure, and endotoxemia.29 How these fatty changes in the liver are induced by a burn injury is largely unknown; however, we posit that hepatic apoptosis (described in detail below) plays a major role in this development. Immediately following the thermal injury, increased hepatic edema is associated with damage to the liver. Jeschke et al.15 have shown that liver and body weight significantly increase at 2-7 days post-burn, as compared to non-burned liver/body weight. We also found that in burned rats, total hepatic protein concentrations were reduced significantly. These findings led to the notion that increased edema, not increased protein levels or hepatocyte numbers, underlies the increase in liver weight. This increase in edema, however, may induce release of hepatic enzymes into the circulation as a result of cellular damage or by altering membrane permeability. Aspartate aminotransferase (AST) and alanine aminotransferase (ALT) are typically detected only at low levels in plasma. Therefore, their detection at elevated levels in the circulation are reliable indications of hepatocyte injury. Because elevations of AST can occur as a result of cardiac arrest or following muscle injury, ALT is considered to be a more specific test for hepatocyte injury. Severe hepatic damage can also be detected by elevations in serum glutamate dehydrogenase or alkaline phosphatase (ALKP). Elevations in ALKP provide insight into the function of the extrahepatic biliary tract as well and are frequently elevated in hepatobiliary disease. The liver damage induced by thermal injury is a result of the combination of edema formation, hypoperfusion, expression of pro-inflammatory cytokines and other cell death signals, and the release of the hepatic enzymes into the circulation. Following a severe burn, elevations between 50% and 200% above age-matched normals are recorded for serum AST, ALT, and ALKP (Fig. 25.2). These serum markers peak early – AST and ALT during the first post-burn day and ALKP during the second,16,26 indicating that burn-induced liver damage is a rapid phenomenon. During the acute hospitalization period, there is regeneration of the liver, which is apparent by the return of these enzymes to baseline. Although these enzymes are elevated as a direct result of the burn injury, additional physiologic processes may be amplifying their expression, including the elevation of ALKP occurring during growth spurts or alongside massive bone resorption. The careful evaluation of hepatic enzyme levels is warranted, and we recommend the comparison of multiple enzymes, as opposed to a single one, to determine whether liver damage is resolving.
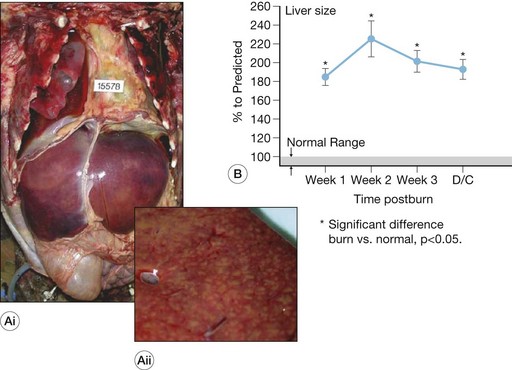
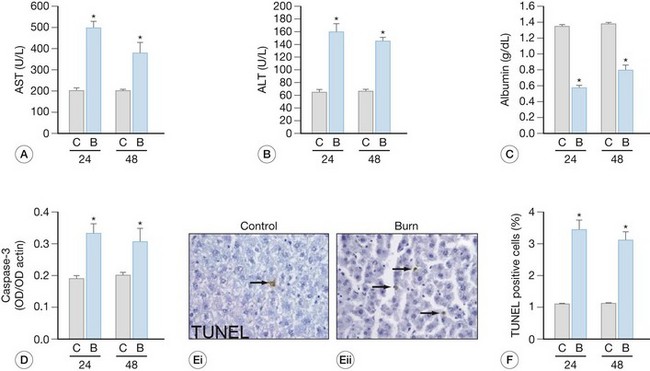
Increases in hepatocyte death, both by apoptosis and necrosis, are associated with liver damage.15 Two distinctly different pathways result in cell death – programmed cell death (apoptosis) and necrosis.30 Cell shrinkage, uniform fragmentation of DNA, and membrane blebbing characterize apoptotic cells, with the cell fragments being phagocytized by neighboring cells. Necrosis, on the other hand, is characterized by cellular swelling, fragmentation of the DNA in a random manner, activation of lysosomes, and complete breakdown of the cellular membrane enabling cellular contents to be extruded into the interstitium. These final steps induce an inflammatory response by attracting inflammatory cells, causing them to release free radicals and pro-inflammatory cytokines, leading to additional tissue breakdown. The morphological hallmarks unique to each process are used to differentiate between apoptotic and necrotic cells. At the time of autopsy, 10–15% of severely burned decedents had liver necrosis, as determined by pathological examination.28 The hepatic necrosis related to burn-induced shock or sepsis was typically focal or zonal, central or paracentral, and sometimes microfocal.
Apoptosis also occurs in the liver following a cutaneous thermal injury (Fig. 25.2).15 The liver tries to maintain homeostasis when hepatocyte apoptosis increases by a compensatory increase in hepatocyte proliferation. Despite the attempt to maintain homeostasis in overall hepatocyte number, the liver is unable to immediately regain mass or maintain protein concentration. A severe thermal injury also can induce apoptosis of the epithelial cells in the small bowel.31,32 Additionally, no increase is found in proliferation of the epithelial cells in the small bowel, resulting in a net loss of mucosal cells, and ultimately this is apparent as reduction in mucosal mass.31,32 Cardiomyocytes are similarly affected by burn injury,33,34 although despite apoptosis of the cells, there is no compensatory increase in cardiocyte proliferation, leading to cardiac impairment and dysfunction.33,34
The molecular mechanisms that initiate and propagate hepatocyte apoptosis following a cutaneous burn are not known, although recent work associates hypoperfusion and ischemia-reperfusion with the induction of programmed cell death.35–38 Blood flow to the bowel is decreased by approximately 60% for up to 4 hours following a thermal injury.31 Hepatic blood flow is likely decreased as well, and this may be one of the early events inducing programmed cell death. Apoptotic signals, including IL-1 and tumor necrosis factor (TNF)-α increase systemically during this same time frame.30,39–42 Additional studies have revealed that these elevations of proinflammatory cytokines are not limited to the serum. Local elevations also occur after a thermal injury, as seen with increased concentrations of hepatic IL-1α, IL-1β, IL-6, and TNF-α.14,43–45 Taken together, these events – reduced splanchnic bloodflow and elevation of pro-inflammatory cytokines – are probably early events in the induction of hepatocyte apoptotic signaling.
Identifying molecular mechanisms
In identifying the molecular mechanisms that mediate the induction of hepatocyte apoptosis and dysfunction following a severe burn (Fig. 25.3), recent work has shown that ER stress is increased following a thermal injury and that cell-wide alterations in calcium are apparent with specific reductions in ER calcium that led to increases in cytosolic calcium.46 Mitochondrial damage occurs as a result of increased cytosolic calcium, leading to the release of cytochrome c which then binds to the inositol triphosphate receptor (IP3R), inducing further depletion of calcium stores in the ER. ER stress and the unfolded protein response trigger apoptosis by activating JNK and initiating the downstream response. The serine on IRS-1 is then phosphorylated, blocking the tyrosine on the same protein from being activated by phosphorylation. At the same time, the pro-survival PI3K/Akt signaling pathway is blocked, amplifying the ER stress response by further activating the IP3R. If the unfolded protein burden can be limited through the use of chemical chaperones, this discovery may be of therapeutic significance as a method to promote hepatocyte survival.47 Alternative pharmacologic agents are being developed to block pro-apoptotic ER stress signaling pathways, and, looking ahead, these alternatives may prove beneficial in the clinic by improving organ function and patient survival.48
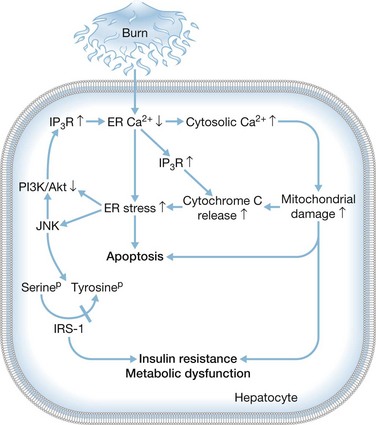
Figure 25.3
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


