Chapter 3
The Head and Neck
- Embryology
- Dental terminology
- Craniofacial surgery
- Cleft lip; cleft lip and palate
- Cleft palate
- Velopharyngeal insufficiency
- Head and neck cancer
- Maxillofacial trauma
- Oculoplastic surgery
- Facial palsy
- Abnormalities of the ear
- Further reading
Embryology
- ‘Branchia’ is the Greek word for ‘gills’.
- Branchial arches are paired swellings along the pharynx of a 4-week-old embryo.
- Humans have six paired branchial arches, but the fifth disappears.
- Each branchial arch contains neural crest cell derivatives:
- A cartilage
- A cranial nerve
- An aortic arch
- Myoblasts.
- First and second branchial arches are most important in facial development.
- Grooves between the arches on their external surfaces are called branchial clefts.
- The cleft between the first and second arch becomes the external auditory meatus.
- The other three clefts disappear.
- Grooves between the arches on their inner surfaces are called pharyngeal pouches, which form:
- First pouch: tubotympanic recess (middle ear, Eustachian tube)
- Second pouch: palatine tonsils
- Third pouch: inferior parathyroids, thymus
- Fourth pouch: superior parathyroids
- Fifth pouch (ultimobranchial body): parafollicular cells of the thyroid.
- Branchial arches are paired swellings along the pharynx of a 4-week-old embryo.
The first branchial arch
- Supplied by the trigeminal nerve and maxillary artery.
- Also known as the mandibular arch, it gives rise to:
- Paired mandibular prominences that contain Meckel’s cartilage.
- This mostly resorbs, but its posterior part forms the malleus.
- The body and ramus of the mandible form from dermal mesenchyme adjacent to Meckel’s cartilage.
- This mostly resorbs, but its posterior part forms the malleus.
- Paired maxillary prominences that form:
- Premaxilla
- Maxilla
- Zygoma
- Squamous portion of the temporal bone.
- Premaxilla
- The quadrate cartilage lies within the maxillary prominence.
- Forms the incus and greater wing of sphenoid.
- Mesenchyme of this arch forms:
- Muscles of mastication
- Anterior belly of digastric
- Mylohyoid
- Tensor veli palatini
- Tensor tympani.
- These are all supplied by the trigeminal nerve.
- Also known as the mandibular arch, it gives rise to:
The second branchial arch
- Supplied by the facial nerve and stapedial artery.
- Also known as the hyoid arch, it contains Reichert’s cartilage, which forms:
- Stapes
- Syloid process
- Lesser cornu and part of the body of the hyoid.
- Stapes
- Mesenchyme of this arch forms:
- Muscles of facial expression
- Posterior belly of digastric
- Stapedius
- Stylohyoid.
- These are all supplied by the facial nerve.
- Muscles of facial expression
The frontonasal process
- Formed by proliferation of mesoderm ventral to the forebrain.
- Not a branchial arch derivative.
- Develops paired placodes (ectodermal thickenings) on its inferolateral borders:
- Medial part of the placode forms the medial nasal process.
- Lateral part of the placode forms the lateral nasal process.
- Medial part of the placode forms the medial nasal process.
- Between the two appears the nasal pit; this becomes the nostril.
- Merging of the medial nasal processes forms:
- The philtrum and Cupid’s bow of the upper lip
- Nasal tip
- Nasal septum
- Premaxilla.
- The philtrum and Cupid’s bow of the upper lip
- The lateral nasal processes form the nasal alae.
Facial development
- Mainly occurs between 4th and 8th weeks of intrauterine life.
- The face is formed from five facial prominences:
- Paired maxillary processes
- Paired mandibular processes
- Frontonasal process.
- Paired maxillary processes
- The medial nasal process fuses with the maxillary process.
- Failure results in cleft lip (CL).
- Bilateral failure of fusion results in a bilateral CL.
- Failure results in cleft lip (CL).
- Failure of midline fusion of the medial nasal processes results in a median CL or Tessier 0 cleft.
- The lateral nasal process fuses with the maxillary process at the alar groove.
- Failure of fusion results in a Tessier 3 cleft.
- Failure of fusion between maxillary and mandibular processes results in macrostomia or Tessier 7 cleft.
- Fibroblast growth factors (FGFs), bone morphogenetic proteins (BMPs), sonic hedgehog (SHH) and retinoic acid have all been implicated.
Cranial development
- Bones of the skull base develop from cartilage precursors, including:
- Sphenoid, ethmoid, petrous temporal and basioccipital.
- Bones of the cranial vault develop in membrane derived from the presumptive dermis, including:
- Frontal, parietal, squamous temporal and squamous occipital.
- At birth, seams of connective tissue called sutures separate the skull bones.
- Where more than two bones meet, there is a wider gap called a fontanelle.
- The posterior fontanelle closes by 3 months.
- The anterior fontanelle normally remains open until 18 months.
- The posterior fontanelle closes by 3 months.
- These fibrous connections allow the skull to deform during childbirth.
- Expansion of the skull is driven by brain growth.
- Brain growth stimulates new bone formation at the suture front.
- Hydrocephalus causes persisting suture patency.
- Microcephaly causes premature suture closure.
- Hydrocephalus causes persisting suture patency.
- The dura plays a key role in controlling suture patency.
The neural crest
- Just prior to fusion of the neural tube, a population of cells known as the neural crest is generated in the area of the neural folds.
- The neural crest contains pluripotential ectomesenchymal tissue.
- Although derived from ectoderm, they exhibit properties of mesenchyme.
- These cells migrate throughout the body.
- They are prevalent within the facial primordia and are essential for normal craniofacial development.
- Teratogens, such as retinoic acid and alcohol, affect neural crest migration.
- Neural crest derivatives encompass:
- The endocrine system, including the adrenal medulla
- The melanocytic system
- Connective tissue, including teeth and bone
- Muscle tissue
- Neural tissue, including the autonomic nervous system.
Developmental terminology and definitions
Malformation
- A morphological defect due to an intrinsic abnormality of development.
- Most common types include:
- Incomplete morphogenesis, such as microcephaly.
- Incomplete closure, such as cleft palate (CP).
- Incomplete separation, such as syndactyly.
- Incomplete morphogenesis, such as microcephaly.
- Malformations initiated earlier in fetal development tend to be more severe.
Deformation
- An abnormality of form or position of a body part due to intrauterine mechanical forces that restrict movement of the developing fetus.
- Deformations can arise from oligohydramnios, bicornuate uterus or twin pregnancy.
- Central nervous system (CNS) malformations can cause deformations due to paralysis.
Disruption
- A defect caused by interference with otherwise normal development.
- In utero amputation of a limb due to an amniotic band is a disruption.
Sequence
- Where a single developmental defect results in a chain of secondary defects.
- Secondary defects may cause further tertiary defects.
- The result is a group of defects traceable to an originating event.
- The primary defect in Pierre Robin sequence (PRS) is mandibular hypoplasia.
- The secondary defect is posterior displacement of the tongue.
- This blocks closure of the palatal shelves resulting in a tertiary defect: CP.
Syndrome
- A group of anomalies (symptoms and signs) containing multiple malformations or sequences.
- Collectively they indicate or characterise a particular syndrome.
- In Greek, syn is with; dromos is running.
Association
- A group of anomalies not known to be part of a syndrome or sequence but found in multiple patients.
- Examples include VATER and CHARGE.
- Not specific diagnoses, but alert clinicians to search for other components of the association.
Dental terminology
- Primary dentition is the first set of 20 ‘baby’ or ‘deciduous’ teeth.
- Permanent dentition refers to the 32 secondary ‘adult’ teeth.
- There are two dental arches: maxillary or upper; mandibular or lower.
- Arches are divided into quadrants by the midline, e.g. maxillary right quadrant.
- Teeth are classified by their morphology:
- Incisors have an incisal edge.
- Canines or cuspids have one pointed cusp.
- Premolars or bicuspids have two cusps.
- Molars have three or more flattened cusps.
- Incisors have an incisal edge.
- Tooth position can be recorded as follows:
- Descriptive, e.g. ‘right upper second molar’.
- Palmer notation, the most popular method in the United Kingdom:
- Teeth are numbered according to their position from the midline.
- Baby teeth are assigned a letter from A to E.
- Addition of a symbol (┘ └ ┐ ┌) indicates the quadrant.
- Teeth are numbered according to their position from the midline.
- The universal numbering system is commonly used in the United States.
- Teeth are numbered from 1 to 32, starting at the right maxillary third molar.
- The maxillary arch is numbered 1–16.
- The mandibular arch is numbered 17–32, starting at the left mandibular third molar.
- Baby teeth are assigned a letter from A to T.
- Teeth are numbered from 1 to 32, starting at the right maxillary third molar.
- Descriptive, e.g. ‘right upper second molar’.
- Teeth have a crown above the gum line and a root below.
- A cusp is a pronounced elevation on the occlusal surface.
- A groove delineates the boundary between adjacent cusps.
- Direction is expressed as follows:
- Buccal: towards the cheek
- Labial: towards the lips
- Lingual: towards the tongue
- Palatal: towards the hard palate
- Mesial: towards the median line, following the curve of the dental arch
- Distal: away from the median line, following the curve of the dental arch
- Apical: towards the apex of the root
- Occlusal: towards the biting surface of a posterior tooth
- Proximal surfaces are those between adjacent teeth.
- Buccal: towards the cheek
- Malocclusion is an incorrect relationship between the teeth of the two dental arches.
- Angle classified malocclusion based on a relationship where the mesiobuccal cusp of the maxillary first molar occludes in the buccal groove of the mandibular first molar.
- Normal occlusion has this molar relationship, with normal alignment of the remaining teeth.
- Class I malocclusion has a normal molar relationship, but there may be overcrowding or misalignment of the other teeth.
- Class II malocclusion has a molar relationship where the buccal groove of the mandibular first molar is distally positioned (away from the median line) from the mesiobuccal cusp of the maxillary first molar.
- Class III malocclusion has a molar relationship where the buccal groove of the mandibular first molar is mesially positioned (towards the median line) from the mesiobuccal cusp of the maxillary first molar.
- Overbite is the amount of vertical overlap of the mandibular anterior teeth by the maxillary anterior teeth.
- Overjet is the horizontal distance between the maxillary incisors and the mandibular incisors.
- Open bite is lack of vertical overlap of the maxillary and mandibular anterior teeth or no contact between the maxillary and mandibular posterior teeth.
- Cross bite is a discrepancy in the buccolingual relationship of the maxillary and mandibular teeth.
- Class I malocclusion has a normal molar relationship, but there may be overcrowding or misalignment of the other teeth.
Craniofacial surgery
Classification
- In 1981, the American Cleft Palate Association published the following classification of craniofacial abnormalities:
- Clefts (centric, acentric)
- Synostoses (symmetric, asymmetric)
- Atrophy–hypoplasia
- Neoplasia–hyperplasia
- Unclassified.
- Clefts (centric, acentric)
- Some conditions fit into more than one category:
- Treacher Collins syndrome is not only associated with facial clefts, but also with facial hypoplasia.
Clefts
- Craniofacial clefts are rare.
- Also known as ‘atypical’ clefts, as distinguished from ‘typical’ CL and palate.
- Occur sporadically once in every 25,000 live births.
- Two leading theories of pathogenesis:
- Classic theory – failure of fusion of the facial prominences.
- Mesodermal penetration theory – lack of mesodermal penetration leads to dehiscence of the epithelial elements.
- Classic theory – failure of fusion of the facial prominences.
- Clefts may also arise from intrauterine compression by amniotic bands.
- Identification of the genetic basis of craniofacial syndromes is a rapidly expanding field.
- Environmental causes include:
- Radiation
- Infections
- Maternal infection with toxoplasmosis, rubella or cytomegalovirus.
- Maternal
- Diabetes, phenylketonuria, maternal age, weight and general health.
- Chemicals
- Folic acid deficiency.
- Vitamin A derivatives, such as isotretinoin.
- Folic acid deficiency.
- Radiation
- Overlap exists between clefts and other hypoplastic syndromes.
- Treacher Collins syndrome is a hypoplastic condition of the lateral face.
- Also known as a confluent Tessier 6,7,8 cleft – has features found in all three cleft patterns.
- Clefts can affect any or all layers of the face.
- They may be unilateral or bilateral.
- Bilateral cases may present different clefts on each side.
- Soft tissue defects do not always correspond to the bony abnormality.
- Craniofacial clefts are often associated with hairline markers.
- These are areas of abnormal linear hair growth along the cleft.
Classification
- Anatomical – Tessier’s classification.
- Embryological – Van der Meulen’s classification.
Tessier’s classification
- The most commonly used and internationally accepted.
- Facial clefts extend downwards from the level of the orbit.
- Cranial clefts extend upwards from the level of the orbit.
- A midline cleft is numbered 0.
- Facial clefts are numbered 1–7.
- Start near the midline with a Tessier 1.
- Each sequential facial cleft is more lateral than the last.
- Tessier 7 is the most lateral, extending outwards from the corner of the mouth.
- Start near the midline with a Tessier 1.
- Cranial clefts are numbered 8–14.
- Tessier 8 is the most lateral and extends into the corner of the orbit.
- Thereafter, each sequential cranial cleft is more medial than the last.
- Tessier 8 is the most lateral and extends into the corner of the orbit.
- Facial and cranial clefts can be connected.
- If this occurs, the patterns tend to add up to 14, e.g. 12 + 2.
- Tessier 30 is a midline cleft of the lower lip and mandible.
- David Fisher, from the Hospital for Sick Children in Toronto, rationalises Tessier’s classification as follows:
- ‘Think in groups of three’:
- 0,1,2: lip to nose
- 3,4,5: lip to orbit/lower eyelid
- 6,7,8: Treacher Collins
- 9,10,11: Orbit and upper lid
- 12,13,14: Medial to orbit.
- 0,1,2: lip to nose
- Tessier 3 involves 4 cavities: oral, maxillary sinus, nasal and orbital.
- Tessier 4 involves 3 cavities: oral, maxillary sinus and orbital.
- Tessier 4 is medial to the infraorbital foramen.
- Tessier 5 is lateral to the infraorbital foramen.
- Tessier 7 is macrostomia.
- Tessier 8 is the ‘equator’.
- Tessier 0,14 is nasofrontal dysplasia.
- Tessier 4 is medial to the infraorbital foramen.
- ‘Think in groups of three’:
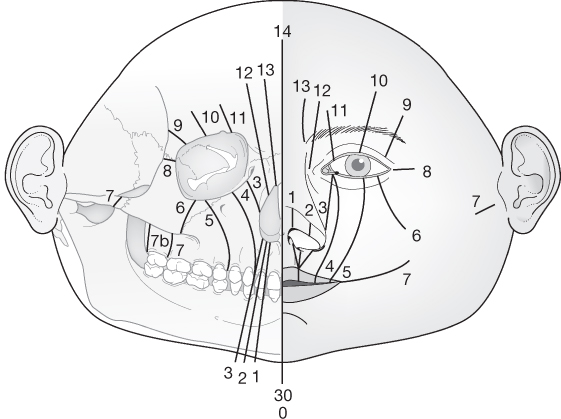
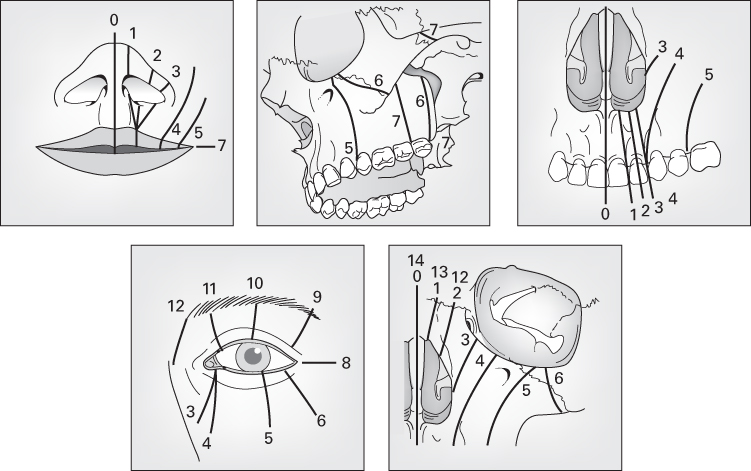
Source: Tessier (1976). Reproduced with permission of Elsevier.
Principles of surgery
- Functions: oral competence, speech and eyelid reconstruction.
- Separation of cavities: oral, nasal and orbital.
- Cosmesis.
- The skeleton can be reconstructed by:
- Removing abnormal elements.
- Transposing skeletal components (including distraction osteogenesis).
- Bone grafting skeletal defects.
- Alloplastic implants.
- Removing abnormal elements.
- Musculature is reattached to the skeleton in its correct anatomical position.
- Sphincters should be recreated where possible.
- Soft tissues can be reconstructed with:
- Local, regional or distant flaps with or without prior tissue expansion.
- Use of Z-plasty to redirect scars.
- Local, regional or distant flaps with or without prior tissue expansion.
- Reconstruction is facilitated by a wide surgical exposure.
- Separation of cavities: oral, nasal and orbital.
Hypertelorism
- An increase in the distance between the bony orbits.
- May be seen in the context of facial clefts.
- The intervening ethmoid sinuses (interorbital space) are overexpanded.
- In practice, it is always a congenital condition.
- Trauma cannot cause true widening of the nasal–orbital walls without creating large midline defects.
- In practice, it is always a congenital condition.
- Hypertelorism may prevent development of binocular vision.
- It is also a significant cosmetic problem.
- Telecanthus is an increase in the intercanthal distance (ICD).
- In telecanthus, the distance between the bony orbits may be normal.
- Pseudotelecanthus is the illusion of telecanthus caused by a flat nasal bridge or prominent epicanthal folds.
Classification
- Tessier graded hypertelorism in adults according to interorbital distance (IOD):
- First degree: IOD 30–34 mm
- Second degree: IOD >34 mm
- Third degree: IOD >40 mm
- First degree: IOD 30–34 mm
- A normal adult IOD is 22–30 mm.
- IOD is measured on a posteroanterior (PA) X-ray or computed tomography (CT) scan as the interdacryon distance.
- The dacryon is the point of union of lacrimal, frontal and maxillary bones.
Causes
- Hypertelorism is associated with numerous conditions:
- Median and paramedian facial clefts
- Sincipital encephaloceles
- Midline tumours.
- Craniofacial syndromes:
- Apert’s
- Crouzon’s
- Craniofrontonasal dysplasia.
- Apert’s
- Median and paramedian facial clefts
Surgical management
- CT is essential for preoperative planning.
- Ophthalmological assessment of visual acuity, amblyopia or extraocular dysfunction is required.
- The orbits can be repositioned without disturbing the optic nerve because the optic foramina are not displaced.
- Tessier gives these basic principles:
- The 360° orbit must be mobilised to allow adequate translocation.
- The ‘functional orbit’ posterior to the equator of the globe must be mobilised.
- A combined craniofacial approach protects the brain.
- The roof of the orbit is also the floor of the anterior cranial fossa.
- The 360° orbit must be mobilised to allow adequate translocation.
Box osteotomy
- Used for patients with hypertelorism and normal midface width.
- Through a coronal approach, osteotomies are made around each orbit.
- Nasal bones and ethmoid sinus are removed, and the orbits moved medially towards each other.
- Alternatively, two paramedian segments are resected, which preserves the nasofrontal junction and cribriform plate.
- Rigid fixation is achieved with plates and screws.
Facial bipartition
- This procedure is well-suited to treat an inverted V deformity of maxillary occlusion.
- Orbital osteotomies leave the floor in continuity with the maxilla.
- Osteotomy through the midincisor line allows medial rotation/transposition.
- An occlusal splint ensures proper positioning of the maxilla.
- Rigid fixation, augmented with bone graft, holds the reduction.
Medial canthopexy
- Osteotomies usually result in detachment of the medial canthal ligament.
- If not reattached, canthal drift gives the appearance of recurrence of the hypertelorism.
Encephaloceles
- Caused by herniation of brain or its lining through a skull defect.
- Frontal skeletal defects can result from Tessier clefts 10, 13 and 14.
- Clinically, they are soft, pulsatile, compressible masses.
- They may transilluminate and have a positive Furstenberg’s sign.
- This is pulsation or expansion of the mass with crying or straining.
- Encephaloceles are classified by their composition into:
- Meningoceles – contain meninges
- Meningoencephaloceles – contain meninges and brain
- Cystoceles – contain meninges, brain and a portion of ventricle
- Myeloceles – contain a portion of spinal cord.
- Meningoceles – contain meninges
- Differential diagnosis includes teratomas, gliomas and dermoids.
- Principles of treatment include:
- Surgical planning aided by ultrasound (US), X-ray, CT and magnetic resonance imaging (MRI).
- Multidisciplinary surgical team for a combined intra- and extra-cranial approach.
- Incision of the sac.
- Amputation of excess tissue to the level of the surrounding skull.
- Dural closure.
- Bony reconstruction.
- Skin closure.
- Surgical planning aided by ultrasound (US), X-ray, CT and magnetic resonance imaging (MRI).
- The remaining intracranial brain tissue should be imaged for abnormalities.
Synostosis
- Premature fusion of one or more sutures in the cranial vault or skull base.
- Occurs approximately once in every 2500 live births.
- May occur:
- As an isolated abnormality or
- As part of a syndrome.
- As an isolated abnormality or
- Nonsyndromal synostosis accounts for 90% of cases.
- Most synostosis syndromes are autosomal dominant.
- The often-cited exception – Carpenter’s syndrome – is autosomal recessive.
- Genetic mutations can be identified in:
- 70% of patients with Crouzon’s, Pfeiffer’s or Saethre–Chotzen syndrome.
- Almost 100% of patients with Apert’s syndrome.
- 70% of patients with Crouzon’s, Pfeiffer’s or Saethre–Chotzen syndrome.
- Craniosynostosis begins during pregnancy or the first year of life.
- Usually complete by 3 years.
Aetiology
- Three theories have been proposed:
- Virchow suggested a primary sutural abnormality.
- McCarthy suggested a dural abnormality.
- Moss suggested abnormality in the skull base.
- Virchow suggested a primary sutural abnormality.
- Many synostotic syndromes may be caused by gene mutations in MSX2, TWIST and fibroblast growth factor receptors 1, 2 and 3 (FGFRs).
- Many craniosynostosis syndromes have associated limb abnormalities.
- Craniofacial and limb development may share common molecular pathways.
Classification
- Synostosis is classified according to:
- The location of the affected suture or sutures.
- The resultant head shape.
- The location of the affected suture or sutures.
- The sagittal suture is the most common single suture synostosis (40–60%).
- It has a male preponderance of 4:1.
- Virchow’s law states that skull growth occurs parallel to a synostosed suture.
- Each pattern of fusion therefore results in a characteristic skull shape:
- Sagittal suture: an elongated keel-shaped skull (scaphocephaly or dolichocephaly).
- One coronal suture: a twisted skull (plagiocephaly).
- Both coronal sutures: a short skull anteroposteriorly (AP) (brachycephaly).
- Compensatory growth may occur upwards (turricephaly, oxycephaly or acrocephaly).
- Metopic suture: a triangular-shaped skull (trigonocephaly).
- One lambdoid suture: a twisted skull (posterior plagiocephaly).
- Both lambdoid sutures: a short skull (brachycephaly).
- Synostosis of multiple sutures leads to a cloverleaf-shaped skull (Kleeblattschädel or triphyllocephaly).
- In Greek, dolichos is long; scaphos is boat; plagios is oblique; brachys is short; acros is high; oxys is sharp; trigonos is triangular; triphyllos is trefoil or three leaves.
- In Latin, turris is tall.
- In German, Kleeblattschädel is cloverleaf skull.
- Sagittal suture: an elongated keel-shaped skull (scaphocephaly or dolichocephaly).
Clinical features
- The most striking feature is abnormal skull shape.
- Because the face is attached to the cranial base, synostosis may limit or distort growth of the face and affect occlusal plane symmetry.
- Midface hypoplasia can lead to obstructive sleep apnoea, requiring continuous positive airways pressure (CPAP) therapy.
- Bicoronal synostosis may result in recession of the fronto-orbital rim.
- Can lead to exorbitism and exposure keratitis.
- Exorbitism is protrusion of the eyeball due to decreased volume of the bony orbit.
- This is different from exophthalmos, which is protrusion of the eyeball due to an increased volume of orbital contents in a normal bony orbit.
- Exorbitism is protrusion of the eyeball due to decreased volume of the bony orbit.
- Can lead to exorbitism and exposure keratitis.
- Patients may show signs and symptoms of raised intracranial pressure (ICP), including:
- Irritability/headaches
- Vomiting
- Tense fontanelles
- Papilloedema
- Developmental delay
- Seizures.
- Irritability/headaches
- Long-term problems of untreated raised ICP include:
- Blindness
- Intellectual disability.
- Blindness
- Incidence of raised ICP increases proportionately with the number of involved sutures:
- Approximately 14% with single suture synostosis.
- Approximately 47% with multiple suture synostosis.
- Approximately 14% with single suture synostosis.
- Normal ICP values in children are arbitrarily set:
- <10 mmHg is considered normal.
- 10–15 mmHg is considered borderline.
- >15 mmHg is considered high.
- <10 mmHg is considered normal.
- Plotting head circumference on a growth chart can help differentiate synostosis from primary microcephaly or hydrocephalus.
- The cranial index (CI) is the ratio of maximum cranial width to maximum cranial length.
- Children with isolated sagittal synostosis typically have CIs of 60–67%.
- Children with normal head shape typically have CIs of 76–78%.
- Children with isolated coronal synostosis typically have CIs of 84–91%.
- CI is not a diagnostic measure but helps quantify the difference between pre- and post-operative head shape.
- The cranial index (CI) is the ratio of maximum cranial width to maximum cranial length.
Radiological features
- Include primary changes in the suture and secondary changes due to abnormal skull growth.
- Primary changes include:
- Loss of suture lucency
- Loss of sutural interdigitations
- Sclerosis of the suture
- Raising (lipping) of the suture.
- Loss of suture lucency
- Secondary changes include:
- Abnormal skull shape
- Harlequin appearance of the lateral orbit on AP films.
- Caused by superior displacement of the lesser wing of sphenoid.
- Copper-beaten appearance of the skull (a sign of raised ICP).
- Widening of the adjacent sutures in compensation.
- Abnormal skull shape
- CT allows direct visualisation of each suture.
- MRI is not used routinely.
- In syndromic cases, it can rule out cerebellar tonsillar herniation (Arnold–Chiari malformation), which may limit compensatory diversion of cerebrospinal fluid (CSF) in raised ICP.
Positional plagiocephaly
- True plagiocephaly is caused by unilateral synostosis of the coronal or lambdoid sutures.
- Positional plagiocephaly is distortion of the skull due to external pressure.
- Incidence of positional plagiocephaly has increased in recent years due to the ‘Back to Sleep’ campaign, which recommends nursing babies supine to reduce the risk of cot death.
- Positional plagiocephaly should be managed nonoperatively, as it is usually self-correcting:
- ‘Active repositioning’ into the prone position during waking hours.
- Special orthotic helmets are commercially available to help remodel the skull.
- Physiotherapy may be required to treat an underlying cause, such as torticollis.
- ‘Active repositioning’ into the prone position during waking hours.
- Clinical features that differentiate positional from true plagiocephaly include:
- Skull shape
- The skull is rhomboid shaped in positional plagiocephaly.
- The occiput is flattened by external pressure, which pushes the ear and forehead forward on that side.
- In true plagiocephaly, one side of the skull does not grow adequately in an AP direction due to synostosis.
- This produces a triangular-shaped skull.
- The base of the triangle lies on the unaffected side.
- The apex of the triangle lies on the affected side, as if pinched at the site of synostosis.
- This produces a triangular-shaped skull.
- The skull is rhomboid shaped in positional plagiocephaly.
- Ear position
- In true plagiocephaly, the distance between the lateral orbit and the ear is asymmetrical.
- Brow shape
- The brow has an ipsilateral prominence in positional plagiocephaly.
- It has a contralateral prominence in true plagiocephaly.
- The brow has an ipsilateral prominence in positional plagiocephaly.
- Cheek position
- The ipsilateral cheek is prominent in positional plagiocephaly.
- Skull shape
Syndromal synostosis
- Synostosis is a feature of over 100 syndromes.
- The more common syndromes share many features:
- Midface hypoplasia
- Skull base growth abnormalities
- Abnormal facies
- Limb abnormalities.
- Midface hypoplasia
Apert’s syndrome
- Occurs once in 160,000 live births.
- Most cases are sporadic but can be autosomal dominant.
- Clinical features:
- Bicoronal synostosis
- Exorbitism with hypertelorism
- Downslanting palpebral fissures
- Midface hypoplasia
- Small, beaked nose
- Class III malocclusion
- CP in 20% of cases
- Complex syndactyly of both hands and feet.
- Bicoronal synostosis
- The hand deformity is classified as:
- Type I: little finger and thumb are separate.
- Type II: only the thumb is separate.
- Type III: all fingers are fused and share a common nail.
- Type I: little finger and thumb are separate.
- There may be nonprogressive ventriculomegaly, but hydrocephalus is uncommon.
- Some have delayed mental development, but many develop normal intelligence.
- Conductive hearing loss is common.
- Cardiac and genitourinary abnormalities found in up to 10% of cases.
Crouzon’s syndrome
- Affects one in 25,000 live births.
- Most are autosomal dominant but can occur sporadically.
- Clinical features:
- Bicoronal synostosis, although other sutures may be involved
- Midface hypoplasia with significant exorbitism
- Normal hands
- High palatal arch; occasional CP
- Class III malocclusion
- Conductive hearing loss.
- Bicoronal synostosis, although other sutures may be involved
- Progressive hydrocephalus, often associated with chronic tonsillar herniation.
- In the absence of raised ICP, developmental delay is rare.
Saethre–Chotzen syndrome
- Estimated to affect one in 50,000 live births.
- Caused by mutations in the TWIST gene (autosomal dominant).
- Clinical features:
- Bicoronal synostosis
- Low-set hair line
- Eyelid ptosis
- Small, posteriorly displaced ears with prominent crura
- Partial syndactyly of the second and third digits is often seen.
- Bicoronal synostosis
- Intelligence is usually normal, but some have mild impairment.
Pfeiffer’s syndrome
- Rare; inherited as an autosomal dominant trait.
- Craniofacial appearance is similar to that of Apert’s.
- A severe form of Pfeiffer’s is characterised by Kleeblattschädel deformity and profound CNS abnormalities.
- Otherwise, intelligence is usually normal.
- Hallmark feature is broad great toes and thumbs.
Muenke’s syndrome
- Discovered in 1996; also known as FGFR3-associated coronal synostosis.
- Differs from other syndromic synostoses because it is defined by a genetic test (Pro250Arg mutation) rather than a constellation of symptoms and signs.
- Affects one in 30,000 and inherited as an autosomal dominant trait.
- Coronal sutures are specifically involved; appearances are otherwise largely normal.
- Hearing loss (30%) and learning difficulties (10%) may occur.
- Reoperation is required more often than with other syndromic synostoses.
Carpenter’s syndrome
- Very rare autosomal recessive condition.
- Various sutures may be involved, commonly sagittal and coronal.
- Intelligence is usually impaired.
- Congenital cardiac abnormalities seen in ⅓ of cases.
- Obesity is typical.
- Clinical features:
- Partial syndactyly of the fingers.
- Preaxial polydactyly of the feet.
- Congenital cardiac abnormalities seen in ⅓ of cases.
Treatment
- Surgery is extensive and potentially risky, associated with significant blood loss.
- Regarded as safe if performed by specialist teams in established centres.
- Main aims of surgery:
- Protect the airway and corneas
- Prevent progressive deformity
- Correct established deformity
- Reduce risks to function from raised ICP.
- Protect the airway and corneas
- Advantages of operating in infancy include:
- Harnessing the most ‘brain push’ to remodel the calvarium during infancy.
- Postsurgical defects ossify more completely before 9–10 months.
- Infantile calvarium is more malleable.
- Harnessing the most ‘brain push’ to remodel the calvarium during infancy.
- However, operating too early can lead to excessive blood loss and difficulties with fixation because the bone is too soft.
- Shunting procedures to treat progressive hydrocephalus are usually done prior to bony remodelling.
- Were both procedures carried out simultaneously, shunting would reduce brain size while vault remodelling would increase skull size.
- The resulting dead space would allow haematoma to collect.
- Were both procedures carried out simultaneously, shunting would reduce brain size while vault remodelling would increase skull size.
- Surgical techniques include:
Sagittal strip craniectomy
- A longitudinal strip of bone over the suture is excised.
Fronto-orbital advancement and anterior cranial vault reconstruction
- The fronto-orbital bar is advanced and held with miniplates.
- This increases the AP dimension of the skull.
- Also deepens the upper orbits, improving exorbitism.
- This increases the AP dimension of the skull.
- The remaining frontal bone is sectioned and replaced in the desired shape.
Posterior cranial vault reconstruction
- Distraction osteogenesis is being used more frequently, given the higher incidence of severe bleeding with posterior vault remodelling.
Le Fort III osteotomy
- Performed to correct midface retrusion encountered in syndromal synostosis.
- May be performed early, aiming to become independent of tracheostomy or ameliorate severe obstructive sleep apnoea.
Monobloc advancement
- Advances the fronto-orbital and Le Fort III segments as one block.
- Associated with higher infection rates and greater blood loss, likely due to communication between cranial and nasal cavities.
Le Fort I osteotomy
- Addresses class III malocclusion and anterior open bite, commonly seen with midface hypoplasia.
- Planned to coincide with completion of orthodontic treatment, at the time of skeletal maturity.
Skeletal distraction
- Many skeletal manipulations are now done by distraction, including:
- Le Fort I
- Posterior cranial vault
- Le Fort III
- Monobloc
- Mandible.
- Le Fort I
- Osteotomies are made as usual.
- A distraction device is fitted either internally or externally.
- As the bone is distracted, the soft tissues are gradually stretched over several weeks.
- Greater advancement can be achieved with distraction than by single-stage advancement.
- Once the desired advancement is achieved, further surgery may be required to stabilise the bones with plates and screws.
- If not, a prolonged period of consolidation is required with the distractor in place.
Post-operative care
- Monitoring of haemodynamic stability and conscious level on a paediatric intensive care unit is required for 24–48 hours.
- A significant amount of swelling is expected.
- Pyrexia of 38 °C for the first 72 hours is not unusual.
Complications
- Overall mortality reported as 1–2% in specialist units.
- Low risk of infection, dehiscence, meningitis, dural tears with CSF leak, air embolus, cerebral oedema and life-threatening bleeding.
- Coagulopathy is most commonly precipitated by massive blood transfusion and low body temperature.
- There may be persisting asymmetry, contour irregularities or incomplete ossification.
Atrophy–hypoplasia
- This group includes:
- Hemifacial microsomia
- Treacher Collins syndrome
- Nager’s syndrome
- Binder’s syndrome
- PRS
- Hemifacial atrophy
- Radiation-induced atrophy–hypoplasia.
- Hemifacial microsomia
- Many of these conditions could be classified as clefts due to the predictable lines of hypoplasia seen in some Tessier clefts.
Hemifacial microsomia
- Congenital condition with a variable phenotype, characterised by underdevelopment of one side of the face. Also known as:
- First and second branchial arch syndrome.
- Otomandibular dysostosis.
- First and second branchial arch syndrome.
- Correlates with a Tessier 7 cleft.
- Hemifacial microsomia is relatively common, affecting one in 4000 live births.
- Unlike Treacher Collins syndrome, it is not usually inherited and is typically asymmetrical.
- It may arise due to haemorrhage from an abnormal stapedial artery.
- Clinical features:
- Underdevelopment of the external and middle ear.
- Underdevelopment of the mandible, zygoma, maxilla, temporal bone, facial muscles, muscles of mastication, palatal muscles, tongue and parotid gland.
- Macrostomia, a first branchial cleft sinus and possible cranial nerve involvement.
- Underdevelopment of the external and middle ear.
- Goldenhar syndrome is a variant of hemifacial microsomia but is typically bilateral, associated with epibulbar dermoids and vertebral abnormalities.
Classification
- The following classifications of hemifacial microsomia have been described:
- OMENS classification
- This classifies deformities of the Orbits, Mandible, Ear, Facial Nerve and Soft tissue.
- Pruzansky classification of the mandible:
- Type I: mild ramus hypoplasia, the body is minimally affected.
- Type IIa: ramus and condyle hypoplasia, but the glenoid–condyle relationship is maintained.
- Type IIb: as for type IIa but with a nonarticulating temporomandibular joint (TMJ).
- Type III: the ramus is very thin or absent with no evidence of a TMJ.
- Mulliken and Kaban subdivided Pruzansky type II as shown.
- Type I: mild ramus hypoplasia, the body is minimally affected.
- Meurman classification of the ear:
- Grade I: small, malformed auricle, but all components present.
- Grade II: vertical remnant of cartilage and skin; atresia of the external meatus.
- Grade III: total or near-total absence of the auricle.
- Grade I: small, malformed auricle, but all components present.
- SAT classification
- Proposed by David, it is analogous to the TNM system used in cancer staging.
- It grades the Skeletal, Auricular and soft Tissue anomalies and suggests a treatment plan.
- Proposed by David, it is analogous to the TNM system used in cancer staging.
Treatment
- Should be individualised to the patient.
- Abnormalities in other organ systems should be sought.
- The following is a general guide:
Before 2 years
- Remove any auricular appendages.
- Correct macrostomia with a commissuroplasty.
- Fit hearing aids.
- Occasional involvement of the fronto-orbital region may require fronto-orbital advancement.
Between 2 and 6 years
- Distraction of the mandibular ramus.
- Pruzansky III deformities may require formal reconstruction of the mandibular ramus.
- This is usually performed with a costochondral rib graft.
Between 6 and 14 years
- Orthodontic treatment.
- Ear reconstruction.
- Soft tissue augmentation:
- Free tissue transfer
- Fat grafting.
- Free tissue transfer
After 14 years
- Augmentation of deficient areas of the facial skeleton.
- Can be done using bone grafts or alloplastic implants, e.g. Medpor®.
- Orthognathic surgery (OGS).
Treacher Collins syndrome
- Also known as:
- Franceschetti syndrome.
- Mandibulofacial dysostosis.
- Tessier 6,7,8 cleft.
- Franceschetti syndrome.
- Autosomal dominant with variable expressivity.
- Affects between one in 25,000 and one in 50,000 live births; equal sex distribution.
Clinical features
- Bilateral symmetrical abnormalities of the first and second branchial arches.
- Characteristic convex facial profile.
- Prominent nasal dorsum and retrusive lower jaw and chin.
- Average or above-average intelligence is the norm.
- Typically, abnormalities are present in the following sites:
Eyes
- Part of the lateral canthus and lower eyelid may be absent (coloboma).
- Eyelashes are often absent medially.
- Atrophic tarsal plate.
- Medially displaced lateral canthus.
- Absent lacrimal apparatus.
- Antimongoloid slant of the palpebral fissure.
- Hypoplastic lateral orbits.
Nose
- Moderately wide bridge with mid-dorsal hump.
- Drooped tip that lacks projection.
Cheek
- The zygoma may be hypoplastic, clefted through the arch or absent.
- A depression may run between the corner of the mouth and angle of the mandible, along the line of a Tessier 7 cleft.
Palate
- CP, with or without CL, occurs in 30% of cases.
- There may be associated choanal atresia.
- If not, the palate is usually high-arched.
Maxilla and mandible
- The ramus of the mandible is often short.
- TMJ and muscles of mastication may be hypoplastic or absent.
- Class II malocclusion with anterior open bite.
Ear
- May be small (microtia) or buried under the skin (cryptotia).
- External meatus may be hypoplastic.
- Middle ear deformities include missing ossicles or cavities.
Cranium
- Reduced cranial base angle (basilar kyphosis).
- Although synostosis is not a feature, the skull may have abnormally short AP length and bitemporal width.
Treatment
Airway
- Patients often have difficulty maintaining their airway due to maxillary and mandibular hypoplasia.
- Tracheostomy may be required.
- Nursing them in a prone position can help improve their oxygen saturation.
- Increased risk of airway obstruction following CP repair.
Feeding
- Factors affecting the airway can also affect swallowing and feeding.
- Failure to thrive requires supplemental tube feeding.
Zygoma and orbit
- Calvarial bone graft can be used to augment the orbital floor and zygoma.
- Lateral canthopexy is done through the same bicoronal incision.
- Usually performed when the child is >7 years, when bony development in that region is almost complete.
Mandible
- Mandibular deformity may be corrected with:
- Rib grafts
- Mandibular advancement
- Bimaxillary procedures
- Le Fort I osteotomy and orthodontic treatment
- Distraction
- Genioplasty
- TMJ reconstruction with costochondral graft (Pruzansky III).
- Rib grafts
- Usually performed at early skeletal maturity, between 13 and 16 years.
Ear
- Reconstruction of the external ear may be required.
- Middle ear surgery is postponed until after auricular reconstruction, to preserve soft tissues.
- Hearing deficits can be improved with hearing aids.
Nose
- Rhinoplasty involves:
- Open approach
- Osteotomies
- Dorsal hump reduction
- Cephalic trim of the lower lateral cartilages
- Columella strut to improve tip projection.
- Open approach
- This is done following any OGS.
Nager’s syndrome
- Also known as acrofacial dysostosis.
- Rare; inherited as an autosomal recessive trait.
- Craniofacial features are similar to Treacher Collins syndrome.
- CP is almost universal.
- There is associated thumb and radial hypoplasia.
- Intelligence is usually impaired.
Binder’s syndrome
- Also known as maxillonasal dysplasia.
- May be inherited as an autosomal recessive trait.
- Clinical features:
- Short nose with flat bridge and short columella
- Absent frontonasal angle
- Absent anterior nasal spine
- Perialar flattening
- Convex upper lip
- Tendency to class III malocclusion.
- May be inherited as an autosomal recessive trait.
Pierre Robin sequence (PRS)
- Affects approximately one in 8500 live births and consists of:
- Micrognathia
- Glossoptosis
- CP.
- Micrognathia
- Due to a small jaw, the tongue occupies a greater proportion of the oropharynx.
- Severe respiratory obstruction can result from the tongue falling backwards (glossoptosis).
- The combined effort of feeding and maintaining the airway is tiring.
- This leads to faltering growth or failure to thrive.
- Most babies have outgrown these difficulties by 6 months, due to mandibular growth and improved neuromuscular control of the tongue.
- Reports suggest underlying genetic mutations in SOX9 and KCNJ2.
Treatment
- Immediate life-saving airway management involves turning the newborn prone to relieve glossoptosis.
- If unsuccessful, a nasopharyngeal airway (NPA) can be inserted to bypass the obstruction caused by the tongue.
- PRS has been classified by severity by the Cleft service at Birmingham Children’s Hospital, UK.
- This classification is also used to guide treatment:
- Grade 1: Nursed side-to-side only.
- Grade 2: Requires nasogastric (NG) feeding and nursed side-to-side.
- Grade 3: Requires NPA, NG feeding and nursed side-to-side.
- Grade 1: Nursed side-to-side only.
- Once stabilised and gaining weight, all PRS babies are discharged with an oxygen saturation monitor.
- A cleft specialist nurse closely monitors weight gain.
- Parents and other carers are trained to manage NG feeds and the NPA.
- Also trained in Paediatric Basic Life Support prior to discharge.
- Birmingham Children’s Hospital successfully treats all PRS babies this way.
- Other more invasive treatments include:
- Glossopexy (tongue–lip adhesion)
- Tracheostomy
- Subperiosteal release of the floor of the mouth
- Distraction osteogenesis of the mandible
- Distraction osteogenesis carries a low but quantifiable mortality rate.
- Distraction osteogenesis of the mandible
- Glossopexy (tongue–lip adhesion)
- Some surgeons maintain that a proportion of severely affected PRS babies require surgical intervention, but that view is controversial.
- PRS may present with other syndromes and anomalies.
- All PRS babies should be screened for Stickler’s syndrome.
- There is a risk of retinal detachment, preventable by early intervention.
- All PRS babies should be screened for Stickler’s syndrome.
Progressive hemifacial atrophy (PHA)
- Acquired condition also known as Parry–Romberg syndrome.
- Usually unilateral; 5% of cases are bilateral.
- Occurs sporadically and not associated with a family history.
- Aetiology is unknown; may be due to viral infection, trigeminal peripheral neuritis or an abnormality in the cervical sympathetic nervous system.
- Presents similar to linear scleroderma in early stages.
Clinical features
- Characterised by gradual wasting of one side of the face and forehead.
- Usually starts between 5 years and late teens.
- Female to male ratio is 1.5:1.
- Involves skin, soft tissue and bone.
- Muscle involvement can also include atrophy of the tongue and palate.
- Typically progresses within the dermatome of one or more divisions of the trigeminal nerve.
- Wasting continues for a number of years before gradually stopping.
- The result is permanent tissue deficiency on one side of the face.
- The following may be seen:
- Localised atrophy of the skin with pigment changes
- Change of hair colour or alopecia
- Change of iris colour and enophthalmos
- A sharp depression on the forehead, occasionally extending into the hairline
- This early sign is known as ‘coup de sabre’
- Atrophy of cheek bone and soft tissue
- Malocclusion.
- Localised atrophy of the skin with pigment changes
Treatment
- Generally, no treatment is performed while the condition is active and progressive.
- Short-term improvements may be achieved with injectable fillers.
- Reconstruction is performed once the condition has been stable for about 12 months.
- This is judged by serial photographs.
- Reconstructive options include:
- Fat and dermofat grafts
- Cartilage grafts
- Le Fort I osteotomy
- Genioplasty
- Alloplastic augmentation
- Temporoparietal fascia and temporalis muscle transfers
- Free tissue transfer.
- Adipofascial flaps are preferred over muscle flaps due to the atrophy of muscle over time.
- Alloplastic augmentation
- Fat and dermofat grafts
Radiation-induced atrophy–hypoplasia
- High dose radiation is used to treat some childhood tumours.
- Deformity may arise by the following mechanisms:
- Impaired growth as the sphenoid ‘locks in’ the upper face.
- The frontal, ethmoid and maxillary sinuses fail to expand.
- Impaired growth as the sphenoid ‘locks in’ the upper face.
- Reconstruction usually involves a combined craniofacial approach:
- Craniotomy to reposition the skull base, orbit and maxilla
- Orbital expansion and mandibular lengthening
- Bone grafts (inlay rather than onlay)
- Free tissue transfer to augment the soft tissues.
- Craniotomy to reposition the skull base, orbit and maxilla
Neoplasia–hyperplasia
Fibrous dysplasia
- Rare, non-neoplastic benign bone disease.
- Lesions are osseous rather than fibrous, characterised by abnormal proliferation of bone-forming mesenchyme.
- Bone maturation stalls at the woven bone stage.
- Usually presents as an enlarging mass in the maxilla or mandible.
- Mass effect can cause cranial nerve compression, proptosis and malocclusion.
- Malignant transformation occurs in 0.5% of patients.
- The condition is usually progressive until about 30 years.
- Diagnosis is confirmed on bone biopsy.
Classification
- Monostotic, with single bone involvement.
- Craniofacially, it usually affects frontal bone, sphenoid or maxilla.
- Can also affect ribs, femur or tibia.
- Craniofacially, it usually affects frontal bone, sphenoid or maxilla.
- Polyostotic, with multiple bone involvement.
- Approximately 3% have the triad of McCune–Albright syndrome:
- Polyostotic fibrous dysplasia
- Precocious puberty
- Café-au-lait macules.
- Polyostotic fibrous dysplasia
- Approximately 3% have the triad of McCune–Albright syndrome:
- Hyperthyroidism and tumours of the pituitary gland may also be found.
- Cherubism is a rare autosomal dominant condition, also known as familial fibrous dysplasia.
- Characterised by multiple areas of fibrous dysplasia within the mandible and maxilla.
- Self-limiting and regresses spontaneously, leaving no deformity.
- Characterised by multiple areas of fibrous dysplasia within the mandible and maxilla.
Treatment
- Early intervention is required when function is at risk:
- Optic nerve compression or other nerve palsies
- Diplopia
- Malocclusion.
- Surgery involves resection of the abnormal areas.
- The resultant defects can be reconstructed with calvarial bone grafts or alloplastic implants.
- Free tissue transfer is used to fill postresection dead space.
- Some cases have responded to bisphosphonate therapy.
- Optic nerve compression or other nerve palsies
Neurofibromatosis
- A group of genetic disorders that predispose to development of Schwann cell tumours.
- Inherited as an autosomal dominant trait with variable penetrance.
- 50% of new cases arise from spontaneous mutations with no family history.
Classification
- Many types have been described; only two forms are commonly encountered:
Neurofibromatosis type 1 (NF1)
- Also known as von Recklinghausen’s disease.
- The most common type (90%), affecting one in 3000 births.
- The mutated gene is on chromosome 17.
- Two of the following clinical features are required for diagnosis:
- Six or more café-au-lait macules (>0.5 cm in children; >1.5 cm in adults)
- Two or more neurofibromas or one plexiform neurofibroma
- Axillary or inguinal freckling
- Optic glioma
- Two or more Lisch nodules (hamartomas of the iris)
- A distinctive osseous lesion with cortical thinning or dysplasia
- A first-degree relative with NF1.
- Six or more café-au-lait macules (>0.5 cm in children; >1.5 cm in adults)
- NF1 is associated with orthopaedic complications, including:
- Scoliosis or kyphosis of the spine
- Congenital bowing and pseudarthrosis of the tibia and forearm.
- Scoliosis or kyphosis of the spine
Neurofibromatosis type 2 (NF2)
- Also known as central neurofibromatosis.
- Affects approximately one in 25,000 live births.
- The mutated gene is on chromosome 22.
- Characterised by bilateral vestibular schwannomas.
- Other abnormalities include:
- Intracranial meningiomas
- Spinal tumours (usually schwannomas or meningiomas)
- Peripheral nerve schwannomas
- Ocular abnormalities (posterior subcapsular lenticular opacities).
- Intracranial meningiomas
Malignancy
- Lifetime risk of malignant transformation is <10%.
- Malignant peripheral nerve sheath tumours (MPNST) arise from Schwann cells.
- Usually occur in plexiform neurofibromas.
- Pain and rapid growth are indicators of malignant degeneration.
- Positron emission tomography (PET) scanning can help localise the malignancy.
- Metastases are common.
- An aggressive surgical attempt at total excision is undertaken.
- 5-year survival following MPNST is approximately 16–52%.
- Malignant peripheral nerve sheath tumours (MPNST) arise from Schwann cells.
Treatment
- Treatment of plexiform neurofibromas requires a multidisciplinary team (MDT) approach.
- Trials are underway looking at imatinib for treatment of plexiform neurofibromas.
- However, surgery is currently the mainstay of treatment.
- The following should be considered:
- Timing and extent of surgery
- Strategies to deal with bleeding refractory to conventional electrocautery:
- Hypotensive anaesthesia, invasive monitoring and cell salvage
- Some advocate packing the wounds open with direct compression.
- Delayed closure then takes place after 48 hours.
- Hypotensive anaesthesia, invasive monitoring and cell salvage
- Timing and extent of surgery
- There is a balance between an acceptable aesthetic outcome and preserving function.
- The limits of surgery should be discussed with the patient.
- Surgery addresses the deformity only; recurrence is usual.
- Improvements are temporary because skin involved with neurofibroma lacks elasticity.
- Specific craniofacial problems associated with NF1 include:
Orbitopalpebral neurofibromas
- Associated with dysplasia of the sphenoid wing.
- Allows the temporal lobe to herniate into the orbit.
- The bony orbit is enlarged, with pulsatile exophthalmos.
- If vision is preserved in the affected eye, the neurofibroma is debulked.
- The orbit can be approached through:
- The upper eyelid for mild lesions
- A bicoronal skin incision with osteotomy into the lateral orbit
- A combined craniofacial approach with frontal craniotomy.
- The upper eyelid for mild lesions
- The sphenoid wing is reconstructed using split rib or calvarial bone graft.
- If vision is impaired, orbital exenteration is performed.
- The eyelid is used for skin cover to allow the orbit to take a prosthesis.
Plexiform neurofibromas of the facial soft tissues
- Cause distortion of the facial soft tissues and skeleton.
- Deformities can lead to visual loss.
- Bony defects are tackled first, with correction of the occlusal plane.
- Soft tissues are reconstructed with free tissue transfer or dermal-fascial-fat grafts.
- Tissue redundancy is addressed through a facelift incision or Weber–Ferguson approach.
- Techniques to minimise recurrence of soft tissue ptosis include:
- Anchoring tissue to bone
- Fascia lata slings.
- Techniques to minimise recurrence of soft tissue ptosis include:
Unclassified craniofacial abnormalities
- These include organ-specific abnormalities:
- Anophthalmia
- Choanal atresia
- Anotia.
- Anophthalmia
Congenital dermoid cysts
- Commonly located in the superolateral orbit, orbital rim and forehead.
- Represent displacement of dermal and epidermal cells into embryonic lines of fusion.
- They differ from inclusion cysts by the presence of dermis and skin adnexa.
- Complete surgical excision is the only effective treatment.
- Can be excised through a supratarsal incision on the upper eyelid.
- Dissection proceeds through the orbicularis oculi onto the cyst.
- Often located deep to periosteum.
- Dissection proceeds through the orbicularis oculi onto the cyst.
Nasal dermoid cysts
- Approximately 10% of dermoids are located on the nose.
- Have a different origin to other dermoids and warrant investigation.
Embryology
- Frontal and nasal bones are separated by a small fontanelle – the fonticulus nasofrontalis.
- A prenasal space exists between the skull base and nasal tip.
- Dura extends through the fonticulus into the prenasal space, where it contacts skin.
- With facial growth, the dura separates from the skin and recedes.
- At this time, the fonticulus nasofrontalis and foramen cecum fuse, forming the cribriform plate.
- Nasal dermoids and sinuses are formed when the dura fails to separate from skin.
- As the dura retracts intracranially, it pulls ectodermal tissue with it.
- Nasal or midline dermoids should be imaged to rule out intracranial extension.
- CT may show a bifid crista galli and patent foramen cecum.
- MRI further demonstrates the extent of intracranial involvement.
- CT may show a bifid crista galli and patent foramen cecum.
Treatment
- A combined craniofacial approach with a neurosurgeon.
- The entire cyst can sometimes be removed through a frontal craniotomy.
- Alternatively, it can be approached by direct incision around the cyst or open rhinoplasty.
- The stalk is traced superiorly, between the cartilaginous septum and nasal bones.
- If the stalk is not excised, craniotomy is required to ensure complete removal.
- Incomplete excision can lead to infection and osteomyelitis.
- The stalk is traced superiorly, between the cartilaginous septum and nasal bones.
Cleft lip; cleft lip and palate
Cleft lip (CL)
- CL is a congenital abnormality of the primary palate.
- The primary palate lies anterior to the incisive foramen and consists of:
- The lip
- The alveolus
- The hard palate anterior to the incisive foramen.
- The primary palate lies anterior to the incisive foramen and consists of:
Cleft palate (CP)
- CP is a congenital abnormality of the secondary palate.
- The secondary palate lies posterior to the incisive foramen and consists of:
- The hard palate posterior to the incisive foramen
- The soft palate.
- The hard palate posterior to the incisive foramen
- Isolated CP is embryologically and aetiologically distinct from CL and CL&P.
Cleft lip and cleft palate
- Both CL and CP may be:
- Complete or incomplete
- Unilateral or bilateral.
- Complete or incomplete
- Unilateral CP occurs when the vomer remains attached to one palatal shelf.
- Bilateral CP occurs when the vomer is attached to neither palatal shelf.
- In isolated CP, the vomer tends to be high and hypoplastic.
Epidemiology
- Incidence of CL&P varies according to the study population.
- In the United Kingdom, CL, CL&P and CP occur once in 700 live births.
- In Caucasians, CL ± CP occur once in 1000 live births.
- In Asia, the incidence is higher: once in 500 live births.
- In Africa, the incidence is lower: once in 2500 live births.
- In the United Kingdom, CL, CL&P and CP occur once in 700 live births.
- Combined CL&P is most common, seen in 50% of cases.
- Isolated CP occurs in 30% of cases.
- Isolated CL occurs in 20% of cases.
- Isolated CP occurs in 30% of cases.
- The ratio of left:right:bilateral CL is 6:3:1.
CL and CL&P
- Twice as common in males.
- Has a familial association.
- The relative risk of a child having CL or CL&P is:
- 0.1% if there is no history of CL or CL&P in the family
- 4% if there is one affected sibling
- 7% if there is one affected parent
- 9% if there are two affected siblings
- 17% if one parent and one sibling are affected.
- 9% if there are two affected siblings
- 0.1% if there is no history of CL or CL&P in the family
- Only 10% of CL and CL&P cases are associated with other abnormalities.
- Van der Woude syndrome is one of the few syndromes associated with CL.
- 1–2% of CL and CL&P patients are affected.
- It has the following characteristics:
- Autosomal dominance
- Lower lip pits
- Absence of second premolar teeth.
- Autosomal dominance
- 1–2% of CL and CL&P patients are affected.
- Environmental teratogens associated with CL and CL&P include:
- Intrauterine exposure to phenytoin (10-fold increased incidence)
- Maternal smoking (twofold increased incidence)
- Maternal diabetes
- Excessive maternal alcohol consumption
- Other anticonvulsants
- Retinoic acid and its derivatives.
- Intrauterine exposure to phenytoin (10-fold increased incidence)
- These are associated, not causal, factors.
- Some studies show that maternal folic acid supplementation produces a lower rate of cleft deformities than predicted.
Isolated CP
- Twice as common in females.
- Fairly consistent worldwide incidence: once in 2000 births.
- Occurs in association with other abnormalities or syndromes in up to 70% of cases.
- Nonsyndromal CP may be associated with the environmental teratogens previously mentioned.
Mechanism
- CL is thought to be caused by either:
- Failure of fusion between the medial nasal process and maxillary process or
- Failure of mesodermal penetration into the layer between ectoderm and endoderm.
- This leads to breakdown of the processes after they have initially fused.
- Failure of fusion between the medial nasal process and maxillary process or
Anatomy
- Complete unilateral CL deformity is characterised by:
- Discontinuity in the skin and soft tissue of the upper lip.
- Vertical and transverse soft tissue deficiency on the cleft side.
- Abnormal attachment of lip muscles into the alar base and nasal spine.
- Outwardly rotated and prominent premaxilla.
- Retropositioned and hypoplastic lateral maxillary element.
- Cleft in the alveolus, usually found at the site of future canine tooth eruption.
- Defect in the hard palate anterior to the incisive foramen.
- Nasal deformity.
- Defect in the hard palate anterior to the incisive foramen.
- Discontinuity in the skin and soft tissue of the upper lip.
Incomplete CL
- CL is incomplete when the cleft does not involve the full height of the lip.
- The nostril sill will therefore be intact.
- Forme fruste, or microform cleft, is a mild form of incomplete CL.
- One or more of the following features may be present in a forme fruste:
- A notch in the vermilion
- A vertical fibrous band from the wet–dry vermilion border (the red line) to the nostril floor
- A kink in the nasal ala on the same side.
- A notch in the vermilion
- Simonart’s band is a soft tissue bridge lying across an otherwise complete CL and alveolus.
- There is no consensus definition; the etymology is interesting.
- Simonart’s band does not convert a complete CL into an incomplete CL.
- There is no consensus definition; the etymology is interesting.
Anatomy of the unilateral CL nasal deformity
- The nasal deformity associated with CL includes:
- Deviation of the nasal spine, columella and caudal septum away from the cleft side.
- Dislocation of the inferior edge of the septum out of the vomer groove.
- Separation of the domes of the alar cartilages at the nasal tip.
- Dislocation of the upper lateral nasal cartilage from the lower lateral cartilage on the cleft side.
- Sagging of the lateral crus of the lower lateral cartilage on the cleft side.
- Retroposition of the alar base on the cleft side.
- Deficient vestibular lining on the cleft side.
- Flattening and displacement of the nasal bone on the cleft side.
- Deviation of the nasal spine, columella and caudal septum away from the cleft side.
Classification
- Popular classification systems include:
Descriptive
- A ‘say-what-you-see’ classification, such as ‘left unilateral complete CL’.
- Can include a schematic diagram showing the relative width and extent of the cleft.
Kernahan’s striped ‘Y’
- A graphical classification, likening the CL&P deformity to the letter ‘Y’.
- Centred on the incisive foramen, dividing primary from secondary palate.
- Each anatomical area is allocated an area on the Y.
- Stippling of a box indicates a cleft.
- Stippling of half a box indicates an incomplete cleft.
- Cross-hatching indicates a submucous cleft.
- The original has been modified by many, including Millard, Jackson and Schwartz, to represent submucous clefts, Simonart’s bands and the nasal deformity.
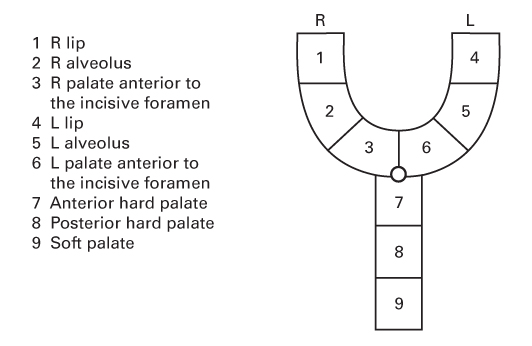
LAHSHAL
- Otto Kriens described this palindromic acronym for clefts.
- Represents the Lip, Alveolus, Hard palate and Soft palate.
- The letters read from the patient’s right to left.
- The second H is sometimes omitted for simplicity.
- Upper case letters represent complete clefts.
- Lower case letters represent incomplete clefts.
- No cleft is represented with a dot.
- An asterisk represents a microform cleft.
- For example:
- . . HSH . . is a complete cleft of the secondary palate.
- l . . . . . . is a right-sided incomplete CL.
- LA . . . AL is a bilateral complete CL and alveolus.
- Represents the Lip, Alveolus, Hard palate and Soft palate.
Veau
- Described in 1931; classifies CL&P into four groups:
- I: Defect of the soft palate alone.
- II: Defect of the hard and soft palate (not anterior to the incisive foramen).
- III: Defects involving the palate through to the alveolus.
- IV: Complete bilateral clefts.
- I: Defect of the soft palate alone.
Organisation of cleft services in the United Kingdom
- Historically, CL&P repair was undertaken by surgeons specialising in plastic, maxillofacial, ENT or paediatric surgery.
- In 1996, UK Health Ministers commissioned a study to advise on standards of care for children with CL&P.
- The Clinical Standards Advisory Group (CSAG) study findings were published in 1998.
- The results were disappointing:
- Many surgeons operated on fewer than 10 cases per year
- Almost 40% of patients had poor or very poor dental arch relationship
- Only 58% of alveolar bone grafts were successful
- 40% of 5-year-old children were in need of treatment for dental caries
- 10% of 12-year-old children had persistent symptomatic oral fistulas.
- Recommendations were based on ‘good practice’ models from Europe and the United States:
- Primary cleft surgeons should see at least 30 new referrals per year
- Expertise and resources should be concentrated from 57 to 8–15 units.
- Cleft surgeons should have undergone extended CL&P training.
- Cleft teams should participate in multicentre audit and research.
- Record keeping should be standardised and protocolised.
- Both child and family should have access to a range of specialties, including paediatrics, clinical psychology and genetics.
- Currently, England and Wales are served by nine different cleft services or networks.
- Scotland, Northern Ireland and the Republic of Ireland have their own services or networks.
- A modern cleft team comprises the following members:
- Cleft coordinator
- Plastic surgeon, maxillofacial surgeon and ENT surgeon
- Cleft specialist nurse, speech and language therapist
- Clinical psychologist, clinical geneticist and audiologist
- Paediatrician, paediatric dentist, orthodontist and medical photographer.
- Parents may require support from cleft specialist nurses regarding:
- Explanation of the diagnosis
- Outline of the likely treatment plan
- Help and advice on how to feed a baby with a cleft
- Psychological and emotional support.
- Many surgeons operated on fewer than 10 cases per year
Timing of repair
- Repair was traditionally performed when the child had attained the three ‘10’s:
- Weight >10 lb
- Age >10 weeks
- Haemoglobin >10 g/dl.
- Weight >10 lb
- There is little firm evidence to support the optimum timing of cleft repair.
- There is little to suggest superiority of neonatal repair.
- Palate closure before 8 years affects maxillary growth; closure after this point does not.
- However, the aim of palate repair is to allow acquisition of normal speech by 5 years.
- This is facilitated by palate repair before speech acquisition begins (with babbling) at 8 months.
- However, the aim of palate repair is to allow acquisition of normal speech by 5 years.
- Options include:
Conventional repair
- Lip and anterior palate repaired at 3 months.
- Any remaining cleft in the secondary palate is repaired between 6 and 12 months.
Delaire technique
- Lip and soft palate repaired simultaneously at 6–9 months.
- Remainder of the palate closed at 14–18 months.
- May result in better midface growth, as less palate dissection is required at the second operation.
Schweckendiek technique
- Soft palate repaired at 6–8 months.
- Lip repaired 3 weeks later.
- Repair of hard palate postponed until 11–13 years.
- Excellent midface growth reported because maxillary growth centres are not disturbed.
- However, only 28% of patients achieved normal speech.
Oslo technique
- Lip repaired at 3 months.
- Anterior palate and alveolar region closed with a vomerine flap during lip repair.
- The vomer flap closes the anterior hard palate and nasal floor in continuity with the lip.
- Remaining palate repaired at 18 months with a modified von Langenbeck repair.
- Critics of the vomerine flap claim the scar at the vomeropalatine suture limits maxillary growth.
Adjuncts to surgery
Presurgical orthodontics
- Presurgical orthodontics involves the application of devices, which:
- Narrow the cleft deformity
- Correct alignment of the alveolar processes
- Mould the nasal deformity.
- Narrow the cleft deformity
- Proponents claim that this:
- Makes subsequent surgical repair easier
- Improves outcome, particularly for the nose.
- Makes subsequent surgical repair easier
- There are two main types of presurgical orthodontic appliances:
- Passive appliances
- Include obturators or feeding plates.
- Prevent displacement of the alveolar arch by reducing distorting forces produced by tongue movement.
- Include obturators or feeding plates.
- Dynamic appliances
- Include the Latham appliance.
- This is pinned into the maxilla intraorally and exerts an active force on the cleft deformity.
- Less invasive alternatives include nasoalveolar moulding.
- Consists of an intraoral plate with attached nasal moulding bulbs.
- Include the Latham appliance.
- Passive appliances
- Not all units utilise presurgical orthodontics; its use is controversial.
- Some reserve it for severe deformities, such as a wide bilateral CL&P.
- There is some evidence that presurgical orthodontics may be detrimental to subsequent growth, although this effect is probably largely related to the Latham.
Lip adhesion
- Essentially converts a difficult wide cleft into a less difficult incomplete cleft.
- Shapes and repositions a protruding premaxillary segment in cases of bilateral CL.
- Can also narrow a wide cleft, facilitating definitive repair.
- May be done at any age, under local or general anaesthesia.
- Skin and mucosal flaps are planned within tissue to be discarded in a definitive lip repair.
- Definitive lip repair is planned 3 months later, after the tissues have softened.
- Disadvantages include possible need for general anaesthesia, additional scar tissue and dehiscence.
- Shapes and repositions a protruding premaxillary segment in cases of bilateral CL.
Techniques of repair
- Principles of management:
- Optimisation of function
- Feeding and growth
- Speech
- Dentition
- Hearing.
- Feeding and growth
- Optimisation of appearance.
- Optimisation of function
- The aims of CL repair are to create:
- A lip that moves normally
- A lip of normal length and width
- Well-aligned anatomical landmarks of the lip:
- Vermilion border, wet–dry mucosal junction (red line), white roll, Cupid’s bow, philtral columns, philtral dimple and nasal sill.
- Symmetry
- Minimal scar.
- A lip that moves normally
- Each technique lengthens the shortened lip on the cleft side, usually by a form of modified Z-plasty.
- The most common techniques are based on either the Millard or Tennison–Randall.
- Whichever technique is used, it is important to perform a functional muscle repair:
- Detach abnormal muscle insertions
- Reconstruct the lip musculature
- The nasalis group of muscles should be attached to the anterior nasal spine
- The orbicularis group of muscles should be attached to each other.
- The nasalis group of muscles should be attached to the anterior nasal spine
- Detach abnormal muscle insertions
Straight-line techniques
- Rose-Thompson
- Mirault-Blair-Brown-McDowell
Upper Z-plasties
- Millard
- Delaire
Lower Z-plasties
- Tennison–Randall
- Le Mesurier (rectangular flaps)
Upper and lower Z-plasties
- Skoog
- Trauner.
The Millard rotation–advancement technique
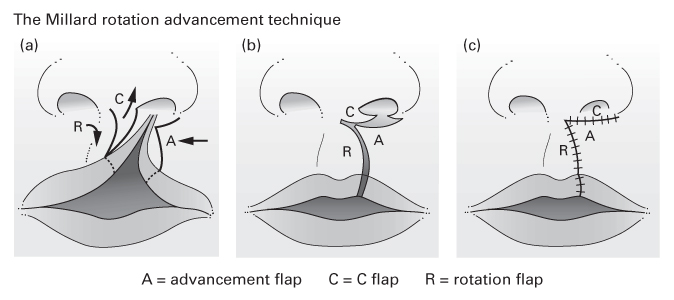
- An upper triangular flap is advanced into the rotation defect of the medial segment.
- Advantages:
- The scar ‘recreates’ the philtral column
- The degree of lip lengthening can be adjusted during surgery
- It has been labelled a ‘cut-as-you-go’ technique.
- Secondary revision is possible by re-elevation and rerotation of the flaps.
- The scar ‘recreates’ the philtral column
- Disadvantages:
- It is a difficult technique to master
- It places a scar across the philtrum at the nasal base
- There is tension at the nostril sill, which can constrict the nostril
- Poor results tend to produce lips that are too short.
- It is a difficult technique to master
The Tennison–Randall technique
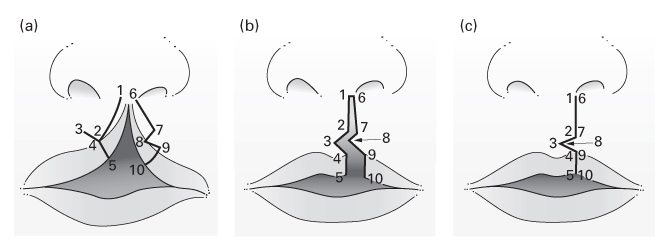
- Described by Tennison; the geometry was elucidated by Randall.
- Advantages:
- Relatively easy to learn
- Maximal tension is below the alveolar ridge, where the lip normally begins to pout.
- Disadvantages:
- Not easy to adjust the degree of lip lengthening intraoperatively
- The philtral column is not restored
- Anecdotally more difficult to revise than a rotation–advancement repair
- Poor results tend to produce lips that are too long.
- Advantages:
How to draw a unilateral rotation–advancement repair
- Practise drawing both right- and left-sided cleft repairs.
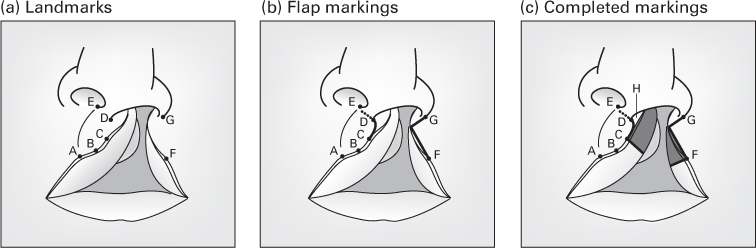
1. Identify the landmarks
- A – peak of Cupid’s bow on the normal side
- B – midpoint of Cupid’s bow, level with the upper lip frenulum
- C – peak of Cupid’s bow on the cleft side, symmetrical with A
- D – columella base on the cleft side
- E – columella base on the normal side
- F – the point where the white roll begins to change (not where it disappears)
- This indicates where underlying muscle insertions become abnormal.
- G – most inferolateral point of the alar base on the cleft side
- Line A–E is the philtral column on the normal side
- This is the normal length of the lip.
- Line C–D is the philtral column on the cleft side
- It is shorter than line A–E.
- The repair must increase distance C–D to match that of A–E.
2. Draw the rotation flap
- The rotation flap is drawn as a curved line between C and D.
- It can be extended along the columella base (dotted line).
- CD is lengthened by straightening the curved line and opening the back-cut at the columella base.
- Some surgeons prefer not to back-cut due to additional scarring.
3. Draw the advancement flap
- Draw a line from G to the vermilion border.
- This line differentiates nasal from facial skin.
- Nasal skin brought onto the face may darken or grow nasal hairs.
- This line differentiates nasal from facial skin.
- The line usually lies perpendicular to the vermilion border.
- A straight line joins this line to point F.
4. Draw flaps for the nostril sill
- A tangent drawn from the rotation flap into the nose delineates Millard’s ‘c’ flap (H).
- This interdigitates with the lip advancement flap and, if done, a vomerine flap.
5. Draw what will be discarded
- Points C and F are extended through the lip perpendicular to the vermilion border.
- The shaded mucosa is excised because it is not required.
- This mucosa would not have existed had the cleft not formed.
- It looks abnormal, and is sometimes called ‘sterile mucosa’.
Primary nasal surgery
- Aims of nasal surgery in CL:
- Restore continuity
- Restore symmetry
- Provide normal function
- Allow normal growth.
- Restore continuity
- Correction of the nasal deformity can be performed at any time:
- Primary surgery, at the time of lip repair
- Delayed surgery, at preschool age
- Late surgery, when facial and nasal growth is complete.
- Primary surgery, at the time of lip repair
- Correction in the late teens usually involves an open rhinoplasty approach.
- The deformity will be improved to some extent by transposing facial muscles into their normal locations during primary CL repair.
- The following techniques have been modified by various surgeons:
McComb technique
- Presurgical orthodontic treatment realigns the skeletal base.
- A ‘hemirhinoplasty’ at the time of lip repair shortens the nose on the cleft side:
- Dorsal dissection between nasal cartilages and skin
- Release of the cleft side alar cartilage from the piriform aperture
- Percutaneous sutures passed through nasal lining into the mobilised alar cartilage, through the dissected subcutaneous space, to exit in the region of the nasion
- The suture is tied over a bolster to lift the alar cartilage into its correct position.
- Dorsal dissection between nasal cartilages and skin
Tajima technique
- Intranasal, reversed U incision to access alar cartilages.
- The incision crosses the alar margin, allowing excision of excess skin causing hooding of the ala on the cleft side.
- The deformed alar cartilage on the cleft side is sutured to three points:
- Ipsilateral upper lateral cartilage
- Contralateral upper lateral cartilage
- Contralateral alar cartilage.
Anderl technique
- The dorsum of the nose is widely undermined to reposition all dislocated structures, including the septum, into their normal position.
- Bony augmentation of the hypoplastic piriform aperture is stimulated by dissecting a fold of mucoperiosteum off the inferior turbinate.
Alveolar bone grafting
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


