Systemic Necrotizing Arteritis: Introduction
|
Introduction
The term “vasculitis” can be defined broadly to mean inflammation of blood vessels. However, from the perspective of the practicing clinician, “vasculitis” is most commonly used to describe a group of diseases in which inflammation of the blood vessels is the major, but not only, pathologic process. The vasculitides are a wide-ranging set of diseases that are mostly idiopathic, rare, and multisystemic. These diseases involve such a variety of clinical presentations and pathologies that all clinicians in every medical and surgical specialty will encounter such patients.
Vasculitis of the skin is a frequent manifestation in many forms of vasculitis, especially small- and medium-vessel arteritis where skin lesions may be the presenting symptom of a systemic illness.
This chapter will focus on skin disease in the systemic vasculitides. Isolated forms of skin vasculitis and some of the systemic vasculitides are covered in Chapter 163. In addition to outlining the skin manifestations of vasculitis in general and for specific types of vasculitis, this chapter will provide an approach to patients with skin disease in which vasculitis is a diagnostic consideration.
Epidemiology of Vasculitis
With the probable exception of drug/toxin-induced vasculitis, all forms of idiopathic vasculitis are considered rare, “orphan” diseases in the United States (prevalences of less than 200,000 people); similar designations exist in Europe and elsewhere.
Vasculitis occurs in people of both sexes, all ages, and all major racial/ethnic groups. However, some forms are more common in certain groups. For example, Takayasu arteritis is substantially more common in women than men, Kawasaki disease is almost exclusively a disease of young children, and giant cell arteritis is limited to older adults. Granulomatosis with polyangilis (GPA) mostly occurs in Caucasians and Behçet disease is markedly more common in countries in the Eastern Mediterranean as well as Japan and Korea. The demographic differences among the vasculitides are of scientific interest as clues to etiology and can be helpful diagnostically.1 However, the epidemiologic tendencies are generally not so strong as to fully exclude the diagnosis of a specific form of vasculitis in any one person and exceptions to the typical epidemiology occur regularly.
Classification of Vasculitis
Multiple systems for classifying vasculitis exist, a situation that reflects a lack of clear understanding of the underlying pathophysiology and the overlap of clinical features among many types of vasculitis.2–7 The most commonly accepted approach to classifying the vasculitides is to sort them by the size(s) of the predominant vessel involved (small, medium, or large) and then subdivide or group diseases, as appropriate (Fig. 164-1).
Figure 164-1
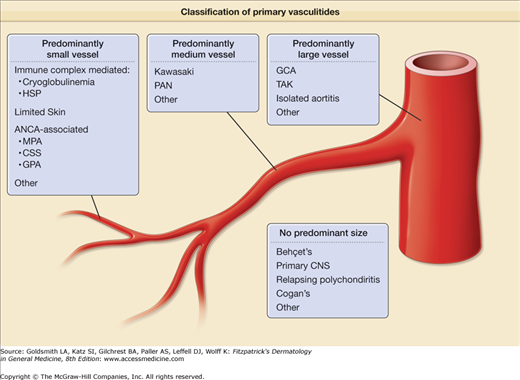
Classification of the primary vasculitides. ANCA = antineutrophil cytoplasmic antibodies; CSS = Churg–Strauss syndrome; GCA = giant cell arteritis; GPA = granulomatosis with polyangiitis (Wegener’s); HSP = Henoch–Shönlein purpura; MPA = microscopic polyangiitis; PAN = polyarteritis nodosa; TAK = Takayasu’s arteritis. (Redrawn from: Watts RA et al: Systemic vasculitis—Is it time to reclassify? Rheumatology (Oxford) Jul 20;2010.)
Classification criteria and definitions have been developed for many, but not all, specific types of vasculitis. These systems were designed for use in clinical research to create fairly homogenous study cohorts and were not meant to be used as “diagnostic” criteria.3 Nonetheless, clinicians will find these criteria helpful. The most widely used criteria for vasculitis are from the classification criteria of the American College of Rheumatology5–6 and the disease definitions of the Chapel Hill Consensus Conference.7 However, these systems do not contain several forms of vasculitis, including some with frequent skin manifestations, such as Behçet disease and cryoglobulinemia, nor any “secondary” vasculitides (associated with another underlying disease such as systemic lupus erythematosus or an infection). Furthermore, some classes are no longer advised for use (e.g., “hypersensitivity vasculitis” is a term that has lost specific meaning). It should be emphasized that the term “leukocytoclastic vasculitis” does not refer to a specific disease but is a pathological description that often, but not always, applies to vasculitis in the skin or other organs. Similarly, “cutaneous vasculitis” is a vague name that could apply to any of several skin lesions seen in vasculitis and is not a distinct entity. There are also some separate sets of criteria for pediatric patients.8
A new international initiative is underway to reconsider the classification of the vasculitides and take into consideration data regarding the clinical and pathophysiological aspects of vasculitis not available when the prior systems were created; such new elements include testing for antineutrophil cytoplasmic autoantibodies (ANCA) and greater availability of advanced imaging techniques for large arterial disease.3
Evaluation of a Patient with Possible Cutaneous Vasculitis
When a patient presents with skin lesions that are concerning for possible vasculitis, answers to three questions should be sought quickly:
Is the lesion due to vasculitis?
Are other organ systems involved in the illness?
Are there additional findings on medical interview, physical examination, laboratory testing, or radiographic imaging that can help establish a specific diagnosis?
If a diagnosis of vasculitis is obtained, then it is imperative to ask two more questions:
Is it possible to make a diagnosis of a specific type of vasculitis for the patient?
Does the patient need immediate treatment and/or hospitalization?
A suggested approach to patients with skin lesions suspected of being due to vasculitis is shown in Fig. 164-2. The answer to the first question is often obtained by a skin biopsy, which is indicated in many cases of palpable purpura or other lesions when a diagnosis of vasculitis is not otherwise easily established. The second question is addressed by a thorough review of systems and physical exam and routine laboratory testing that can usually be completed rapidly. The third question is addressed by more specialized laboratory tests for which results typically take several days to return. It is important to quickly identify organ system involvement and how “sick” the patient is (or might soon be), since some causes of cutaneous vasculitis require no treatment, but others require immediate hospitalization for initiation of immunosuppressive and supportive therapy.
Figure 164-2
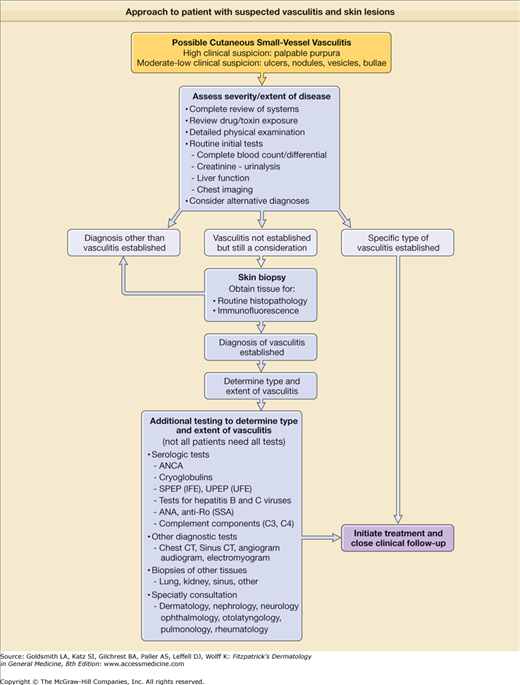
Approach to the patient with suspected vasculitis and skin lesions. ANCA = antineutrophil cytoplasmic antibodies; ANA = antinuclear antibodies; CT = computed tomography; IFE = immunofixation electrophoresis; SPEP = serum protein electrophoresis; SSA = susceptible S. aureus; UFE = uterine fibroid embolization; UPEP = urine protein electrophoresis.
A full review of systems with assessment of the overall severity of illness is the single most important component of the early evaluation of a patient suspected of having vasculitis. Together, the diseases that cause cutaneous vasculitis can affect all organ systems and, in most cases, that involvement will cause symptoms—renal disease being a prominent exception. Although some symptoms are clearly more concerning than others (hemoptysis vs. dry cough, painful red eye vs. mild arthralgias), even relatively mild symptoms can be a clue that disease is not limited to the skin. A list of important signs and symptoms of vasculitis is shown in Table 164-1.
Organ System | Signs and Symptoms | Disease Process | Type of Vasculitis |
|---|---|---|---|
| General | |||
| Fever | Systemic inflammation | Many of the systemic vasculitides | |
| Fatigue/malaise, | Systemic inflammation | Most of the systemic vasculitides | |
| Weight loss | Systemic inflammation | Most of the systemic vasculitides | |
| Eye | |||
| Red eye | Episcleritis, scleritis, uveitis, conjunctivitis | GPA, MPA, RPC, BD, others | |
| Acute visual loss or amaurosis fugax | Arterial insufficiency | GCA, TAK | |
| Proptosis | Orbital granuloma (“pseudotumor”) | GPA | |
| Tearing | Dacrocystitis with lacrimal duct occlusion | GPA, CSS | |
| Ear | |||
| Hearing loss | Sensorineural hearing loss | GPA, MPA, CSS, GCA | |
| Conductive hearing loss | GPA, CSS, RPC | ||
| Ear pain and fullness | Mastoiditis and/or other inflammation of upper airway, auditory tube, middle ear | GPA, CSS | |
| External ear redness, tenderness, swelling | Chondritis | RPC, GPA, CSS | |
| Nose and sinuses | |||
| Epistaxis, nasal crusting and discharge | Nasal mucosal inflammation | GPA, CSS | |
| Nasal bridge collapse (saddle nose deformity) | Nasal cartilage inflammation | GPA, RPC | |
| Nasal polyps | Eosinophilic nasal inflammation | CSS | |
| Facial pain/tooth pain | Sinusitis | GPA, CSS | |
| Anosmia | Olfactory epithelium/cells damage | GPA, CSS | |
| Oral cavity | |||
| Painful oral ulcers | Aphthous ulcers | BD, GPA | |
| Gingival pain and swelling | Gingival inflammation | GPA | |
| Jaw claudication | Arterial insufficiency to muscles of mastication | GCA | |
| Pulmonary | |||
| Hemoptysis | Alveolar hemorrhage | GPA, MPA | |
| Pulmonary artery rupture | BD | ||
| Pulmonary embolus | GPA, MPA, CSS, BD | ||
| Pulmonary nodules | GPA, CSS | ||
| (See also causes of “Hemoptysis”) | |||
| Dyspnea/cough | Pulmonary infiltrates | GPA, MPA, CSS | |
| Bronchitis/large airway collapse | GPA, RPC | ||
| Pleuritis | GPA, CSS | ||
| Subglottic stenosis | GPA, RPC | ||
| Stridor/dyspnea | Asthma | CSS | |
| Wheezing | Large airway collapse | GPA, RPC | |
| Cardiovascular | |||
| Angina | Coronary arteritis | TAK | |
| Congestive heart failure | Aortic root/valvular disease | TAK, GCA, BD, RPC | |
| Myocarditis | CSS, GPA | ||
| Limb claudication | Aortic valve insufficiency | TAK, GCA | |
| Large artery stenosis | TAK, GCA | ||
| Gastrointestinal | |||
| Abdominal ischemic pain | Arterial insufficiency | PAN, HSP, GCA, TAK | |
| Lower GI bleeding | Muscoal ulcers or infarction | HSP, CSS, GPA, MPA, PAN | |
| Renal | |||
| Gross hematuria | Renal infarction | PAN | |
| Glomerulonephritis (rare cause of gross hematuria) | GPA, MPA, CSS, HSP, Cryo | ||
| Central nervous system | |||
| Headache, scalp tenderness | Cranial arteritis | GCA, TAK | |
| Lightheadedness/syncope | Arterial insufficiency to brain | GCA, TAK | |
| Cranial neuropathy | Inflammation of nerves; rarely mass lesion | GCA, GPA, MPA | |
| Peripheral nervous system | Sensory/motor dysfunction | Inflammation of nerves; rarely mass lesion | CSS, GPA, MPA, PAN, Cryo |
| Musculoskeletal | |||
| Polyarthralgia | Polyarthritis | GPA, GCA, TAK, Cryo, HSP, BD | |
| Shoulder and hip girdle pain | Polymyalgia rheumatica | GCA | |
| Muscle weakness | Myositis | CSS | |
| Skin | |||
| Purpura | Small-vessel vasculitis | GPA, MPA, CSS, PAN, HSP, Cryo | |
| Painful nodules, deep ulcers | Medium-vessel vasculitis | PAN, GPA, MPA, Cryo | |
| Digital ischemia/gangrene | Medium-large artery stenosis | GCA, TAK, PAN, GPA, MPA, Cryo | |
| Superficial nodules | Granulomas | GPA, CSS | |
| Papules, acne-like lesions | Papulopustular lesions | BD | |
| Painful, red nodules | Erythema nodosum | BD, TAK | |
| Peripheral edema | Deep vein thrombosis | GPA, MPA, CSS, BD |
It is critical to know the full medical history of any patient suspected of having vasculitis. Other diseases may either have vasculitis as a component of the illness (e.g., lupus) or may cause skin lesions that mimic vasculitis. Drug-induced vasculitis (DIV) is common and skin lesions, usually but not always purpura, are the most common manifestation of DIV.9 The list of drugs reported to cause vasculitis is enormous with almost every class of medication implicated in possible cases of DIV. It is useful to ask about prescription, nonprescription, and “alternative” or herbal mediation use in the prior 6–12 months since the effect of some medications may persist after usage ends. Patients should also be asked about use of illegal or recreational drugs since several such agents, including methamphetamines, cocaine, and others have been implicated in cases of vasculitis. Occupational or other exposure to nondrug toxins should be asked about.
The patient should be asked about not only usual signs and symptoms of infection, but also recent travel, contacts with sick individuals, and risks for sexually transmitted diseases.
Beyond a careful and full assessment of the skin, a multisystem examination is useful to determine whether symptoms are associated with objective abnormalities, or whether there are findings that a patient has not noticed. Vital signs are essential, but a patient with normal blood pressure can still have severe glomerulonephritis. The eyes should be inspected for redness and proptosis. The anterior nasal cavity can be easily visualized with an otoscope. Evidence of lymphadenopathy should be sought. Cardiac, lung, and abdominal examinations can give clues to underlying disease, but normal examinations do not rule out pathology. Similarly, absent pulses, asymmetric blood pressure readings, and bruits are helpful but imperfect measures to screen for large-vessel vasculitis. A complete joint examination is important and any findings suggestive of synovitis (joint swelling, warmth, redness) must be further investigated; however, many patients with vasculitis will have arthralgias without joint effusions. A full neurological examination is one of the most valuable components of the evaluation and triage of a patient suspected of having vasculitis; subtle sensory and even motor abnormalities may be missed on initial evaluation.
The more detailed and expert examinations that can be performed by ophthalmologists and otolaryngologists are often extremely helpful in evaluating patients suspected of having vasculitis. Urgent referral is often indicated in patients with concerning symptoms such as new visual impairment, painful or red eyes, hoarseness or stridor, or hearing loss.
Given the broad range of entities that fall under the category “vasculitis”, and the even larger number of diseases that are also reasonably considered when evaluating a patient suspected of having vasculitis, a vast number and range of diagnostic tests are often considered in such cases. However, obtaining a thorough medical history and conducting a detailed physical examination should enable the clinician to limit the types of vasculitis under consideration and prioritize the ordering of diagnostic tests. Not all tests need be ordered for all patients suspected of having vasculitis. This approach must of course be balanced by the possibility of atypical presentations of vasculitis as well as a range of infections, malignancies, and other diseases in the differential diagnosis of such patients. Evaluation for possible vasculitis usually occurs in parallel to evaluation for other processes.
The method for diagnosing vasculitis depends on the type of vasculitis suspected, which is often based on the size of vessel involved. Vasculitides affecting the skin usually involve small- and medium-sized vessels and these vessels are amenable to biopsy (Fig. 164-3). Given the ease and low risk of skin biopsies, they play an important role in diagnosing vasculitis, and an equally important role in establishing a diagnosis other than vasculitis. A standard punch biopsy is sufficient to diagnose small-vessel vasculitis but a deeper and wider excision may be necessary to capture information on medium-sized vessels.10,11 Lesions that should be approached with a deeper biopsy include subcutaneous nodules, livedo reticularis, or deep ulcers (Fig. 164-4). Many types of small-vessel vasculitis may also involve medium-sized skin vessels. It is important to realize that the difference between “small” and “medium” vessels is somewhat subjective and skin pathologists make such distinctions more often than might other pathologists who see larger biopsy specimens.
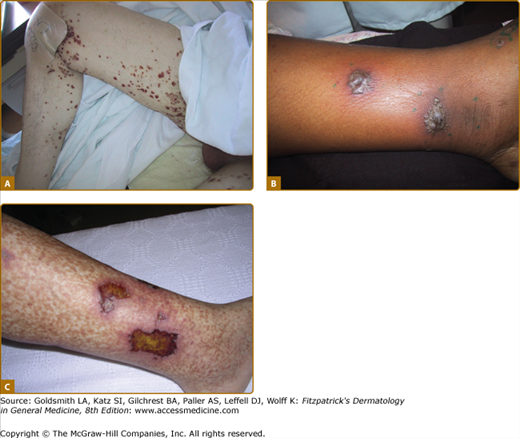
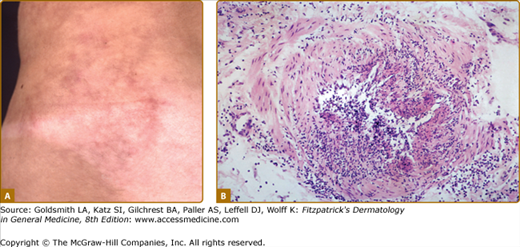
Sometimes the presence of a typical clinical syndrome makes biopsy unnecessary. For example, Henoch–Schöenlein purpura in children is often diagnosed on clinical grounds alone and some cases of ANCA-associated or cryoglobulinemic vasculitis can be diagnosed confidently by combining clinical features with specific serologic tests. Behçet disease and Kawasaki disease are diagnosed based on the clinical syndromes; biopsy is usually not performed on the skin lesions that are common in these diseases, and such biopsies are often nondiagnostic.
It is generally recommended to biopsy a skin lesion that has been clinically apparent for less than 48 hours, if possible, to maximize the chance of finding the typical features of acute neutrophilic vasculitis, including fibrinoid necrosis, extravasation of erythrocytes, extravasation of neutrophils with release of nuclear debris (leukocytoclasia), and the presence of immune deposits.10,11 Processing of tissue is different for conventional histopathology or immunofluorescence testing; if immunofluorescence is desired, then either two specimens need to be obtained, or a single specimen needs to be divided before processing. The latter approach may however damage the tissue.10
As discussed throughout this chapter, the histologic finding of leukocytoclastic vasculitis is helpful in confirming the diagnosis of vasculitis but does nothing to establish an etiology from among the broad number of possibilities. Microscopy sometimes reveals features that are suggestive but not diagnostic of vasculitis, such as leukocytoclasia without fibrinoid necrosis. The finding of a perivascular infiltrate, particularly if it consists predominantly of mononuclear cells but even if it is neutrophilic, is also nonspecific. Certain features, when seen in addition to leukocytoclastic vasculitis, are strongly suggestive of particular diseases, such as extravascular granulomas with geographic necrosis (GPA), or eosinophil-rich extravascular granulomas (Churg–Strauss syndrome; CSS),12 but these features are seen in a minority of biopsies in these diseases.
A predominance of IgA over IgG/IgM by immunofluorescence is suggestive but not diagnostic of Henoch–Schönlein purpura. The presence of deposits of IgG, IgM, and/or complement is suggestive of one of several immune-complex-mediated etiologies, including drug hypersensitivity, postinfectious vasculitis, cryoglobulinemia, and vasculitis secondary to systemic lupus erythematosus, Sjögren syndrome, or rheumatoid arthritis.12
Vasculitis is often diagnosed by biopsy of other organs, such as kidney, lung, muscle, or peripheral nerve or even from surgical specimens (Fig. 164-5). Kidney or lung biopsies are more likely than skin biopsies to show pathology diagnostic of a particular disease. Nonetheless, a skin biopsy establishing the diagnosis of vasculitis may preclude the need for more invasive biopsies.
Figure 164-5
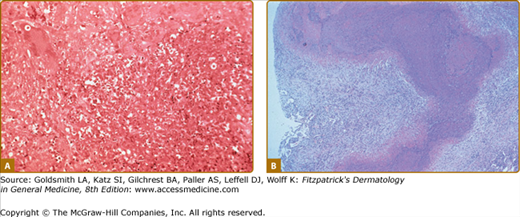
A. Lung histopathology from a patient with granulomatosis with polyangiitis (Wegener’s) demonstrating necrosis, giant cells, and mixed cellular inflammation. B. “Geographic necrosis” in a low-power view of an open lung biopsy specimen from a patient with granulomatosis with polyangiitis (Wegener’s).
Although individual laboratory tests on their own are almost never diagnostic for vasculitis, such tests are essential in the evaluation of a patient in whom cutaneous vasculitis is being considered. Laboratory testing may identify organ systems involved in the disease process, especially renal disease. Furthermore, in the proper setting, selected serologic tests may establish an etiology for vasculitis. However, serologic tests usually complement rather than substitute for biopsy, particularly in a patient with skin lesions that can be readily biopsied.
Tests for renal disease are the most important laboratory tests to order in evaluating a patient suspected of having vasculitis since renal disease is common in many vasculitides and is rarely accompanied by signs or symptoms until end-stage renal failure occurs. Urinalysis, including both dipstick and microscopic examinations, should be performed on all patients in whom vasculitis is suspected, and repeatedly in patients in whom vasculitis of small- or medium-sized vessels is established in another organ system. The presence of any blood on the routine dipstick tests needs to be followed by an examination for red blood cell casts by someone specifically trained to look for casts (many nephrologists, some rheumatologists, but few laboratory technicians in North America). Measurement of serum creatinine is critical to estimate the glomerular filtration rate (GFR). Small changes in creatinine, even within the normal range, may be early evidence of decline in GFR. Although small-vessel vasculitis affecting the glomeruli is expected to produce hematuria, usually accompanied by red blood cell casts and proteinuria, vasculitis affecting only medium-sized vessels (e.g., polyarteritis nodosa) typically produces either isolated hematuria or a normal urinalysis. Urinalysis and serum creatinine are equally important tests and are complementary; neither alone is sufficient to exclude renal disease in vasculitis.
Vasculitis, particularly polyarteritis nodosa, can involve the liver, but significant hepatic dysfunction is rare. Liver function tests are thus of limited value in diagnosing vasculitis, but they do provide a baseline against which future values can be compared if, as is often the case, potentially hepatotoxic drugs are to be used for treatment. Liver function tests can also provide an early hint at infection with hepatitis B or C viruses, both of which are associated with vasculitis, but do not substitute for serologic testing for these infections. Normal liver function tests do not rule out infectious hepatitis.
A complete blood count should be ordered on all patients suspected of having vasculitis. Many patients with active vasculitis have anemia and/or thrombocytosis, but the same is true of a wide range of inflammatory diseases. Severe anemia can be a clue to serious gastrointestinal involvement from various forms of vasculitis. The white blood cell count and differential can also be clues to the presence of infection or hematologic malignancy. However, leucocytosis is usually nonspecific and is also commonly caused by use of glucocorticoids. An elevated absolute eosinophil count is found in most untreated patients with CSS and a count greater than 1,000 cells/μL helps differentiate this disease from asthma and atopy.
The erythrocyte sedimentation rate (ESR) and levels of C-reactive protein (CRP) are elevated in many patients with vasculitis, but the diagnostic sensitivity and specificity of these tests are not particularly high and thus these tests are not particularly helpful in either establishing or excluding a diagnosis of vasculitis. Furthermore, the levels of ESR and CRP do not correlate well with stage or severity of disease. ESR and CRP are often elevated in conditions that mimic vasculitis in the skin, as well as in many serious systemic diseases, including infections and malignancies. Patients with active vasculitis can have normal ESR and CRP values and patients may remain in clinical remission despite persistent elevation of these markers after treatment.
Testing for autoantibodies is often a critical component of establishing the type of vasculitis present but it is important to recognize that serologic testing on its own is never diagnostic and should never substitute for clinical judgment.
Testing for ANCA and antiglomerular basement membrane (anti-GBM) antibodies, as well as antinuclear antibodies (ANA) to address the alternative possibility of systemic lupus erythematosus, is advised for any patient presenting with pulmonary hemorrhage and/or acute renal insufficiency with an active urinary sediment. ANCA-associated vasculitis and lupus can present with vasculitis of the skin, but anti-GBM disease does not, so the latter topic will not be discussed further.
Approximately 90% of patients with microscopic polyangiitis, 75% of patients with GPA, and 40% of patients with CSS will test positive for ANCA.13–16 Modern ANCA testing includes both immunofluorescence staining of neutrophils for the cytoplasmic (c-ANCA) or perinuclear (p-ANCA) patterns and ELISAs for specific autoantigens [proteinase-3 (PR3) and myeloperoxidase (MPO)].16–18 Specificity of positive testing for anti-PR3 and anti-MPO antibodies for ANCA-associated vasculitis is quite high,17–19 but specificity of p-ANCA staining in the absence of anti-MPO antibodies is low. Thus, positive tests for ANCA by ELISA are essential to consider ANCA testing positive for purposes of diagnosing vasculitis.
The predictive value of positive ANCA testing depends on the setting. In cases of biopsy-proven vasculitis or clinical “surrogates” of a biopsy of vasculitis, such as diffuse alveolar hemorrhage or acute renal failure with an “active” urinary sediment, positive testing for anti-PR3/MPO ANCA is highly specific. In the setting of nonspecific constitutional and musculoskeletal symptoms, the positive predictive value of ANCA testing is lower.
Testing for ANA and related autoantibodies is useful when there is suspicion of systemic lupus or Sjögren syndrome. ANA testing is extremely sensitive (>95%) but not specific for the diagnosis of lupus. With the exception of anti-Ro (SSA) antibodies, additional tests for specific nuclear antigens, including double-stranded DNA, Smith, RNP, and La (SSB), should only be ordered if the ANA is positive and lupus is still under consideration. Only 80% of patients with Sjögren syndrome test positive for rheumatoid factor (RF), anti-Ro (SSA), or anti-La (SSB) antibodies, so negative tests do not rule out this diagnosis.
Testing for rheumatoid factor is rarely useful in establishing either the diagnosis or specific type of vasculitis. The sensitivity and specificity of rheumatoid factor for Sjögren syndrome or cryoglobulinemic vasculitis are low. While at least 70% of patients with rheumatoid arthritis test positive for rheumatoid factor, the test is positive in more than 95% of patients with rheumatoid vasculitis.20 However, since rheumatoid vasculitis typically occurs in patients with longstanding, severe rheumatoid arthritis, such testing has little additive value.
Cryoglobulins are immune complexes (immunoglobulins and their target antigens) that precipitate in the cold and are associated with clinical syndromes in which vasculitis is a prominent component (see Chapter 169). Cryoglobulinemia most commonly results from chronic infection with hepatitis C virus, but rheumatoid arthritis, systemic lupus erythematosus, Sjögren syndrome, and hematologic malignancies are also all associated with cryoglobulinemia. Testing for cryoglobulins requires careful attention to specimen handling and processing since incorrect practice at any one of several steps results in a high false-negative rate. Similarly, standard serum protein electrophoresis testing may not pick up some immunoglobulin clones, and immunofixation electrophoresis is a more comprehensive screen for clonal immunoglobulins.
Vasculitis has also been associated with monoclonal gammopathies (myeloma, plasmacytoma, or lymphoma) in the absence of cryoglobulinemia.21
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


