Systemic Glucocorticoids: Introduction
|
Glucocorticoids (GCs) are a mainstay of dermatologic therapy because of their potent immunosuppressive and anti-inflammatory properties. In 1949, Hench and coworkers1 described the beneficial effects of cortisone in patients with rheumatoid arthritis. By understanding the properties and mechanisms of action of glucocorticoids, one can maximize their efficacy and safety as therapeutic agents.
Mechanism of Action
The major naturally occurring glucocorticoid is cortisol (hydrocortisone). It is synthesized from cholesterol by the adrenal cortex. Normally, less than 5% of circulating cortisol is unbound; this free cortisol is the active therapeutic molecule. The remainder is inactive because it is bound to cortisol-binding globulin (CBG, also called transcortin) or to albumin. The daily secretion of cortisol ranges between 10 and 20 mg, with a diurnal peak around 8:00 am.2 Cortisol has a plasma half-life of 90 minutes. It is metabolized primarily by the liver, although it exerts hormonal effects on virtually every tissue in the body. The metabolites are excreted by the kidney and the liver.
The mechanism of glucocorticoid action involves passive diffusion of the glucocorticoids through the cell membrane, followed by binding to soluble receptor proteins in the cytoplasm.3 This hormone-receptor complex then moves to the nucleus and regulates the transcription of a limited number of target genes. There are three main mechanisms of glucocorticoid action. The first is direct effects on gene expression by the binding of glucocorticoid receptors to glucocorticoid-responsive elements, leading to the induction of proteins like annexin I and MAPK phosphatase 1. Annexins reduce phospholipase A2 activity, which reduces the release of arachidonic acid from membrane phospholipids,4 limiting the formation of prostaglandins and leukotrienes.5,6 The second mechanism is indirect effects on gene expression through the interactions of glucocorticoid receptors with other transcription factors. Some of the most important appear to be inhibitory effects on the transcription factors AP-1 and NF-κB, coupled with increased IκB, an inhibitor of NF-κB,7 This decreases the synthesis of a number of proinflammatory molecules, including cytokines, interleukins, adhesion molecules, and proteases. The third is glucocorticoid-receptor-mediated effects on second messenger cascades through nongenomic pathways such as the PI3K-Akt-eNOS pathway.8,9
The human GC receptor (GR) messenger RNA has alternative splice variants, glucocorticoid receptor–α and –β. The relative levels of these two variants influence the cell’s sensitivity to glucocorticoid, with higher levels of GR-β being one of many mechanisms leading to glucocorticoid resistance.9,10
There is usually a delay in the onset of pharmacologic activity of glucocorticoids relative to their peak blood concentrations, which is probably consequent to altering the transcription of genes,8 although some actions appear to be independent of transcription.11 Glucocorticoids may also exert their effects by nongenomic mechanisms such as membrane-bound receptors and/or physicochemical interactions with cellular membranes.12 Some effects of glucocorticoids are too rapid to be mediated by genomic glucocorticoid action.13 This mechanism might explain the additive benefits of very high-pulse glucocorticoids.
The recognition that desired clinical effects of GC treatment on the GR are mediated by transrepression mechanisms, while transactivation mediates side effects, have led to the development of compounds—selective GR agonists (SEGRAs)—that potentially will give glucocorticoid effects without the same amount of toxicity.8,14,15 The multiplicity of biologic effects produced by glucocorticoids emphasizes that currently there is no unifying hypothesis to explain the therapeutic efficacy of these extremely potent anti-inflammatory and immunosuppressive agents.
Cellular Effects of Corticosteroids
Glucocorticoids profoundly affect the replication and movement of cells. They induce monocytopenia, eosinopenia, and lymphocytopenia and have a greater effect on T cells than on B cells.16 The lymphocytopenia appears to be caused by a redistribution of cells as they migrate from the circulation to other lymphoid tissues, and it has been suggested that glucocorticoids induce apoptosis.17 The increase in circulating polymorphonuclear leukocytes is related to demargination of cells from the bone marrow and a diminished rate of removal from the circulation, at least partially mediated by the increase in annexin 118; there also appears to be inhibition of neutrophil apoptosis.19
Glucocorticoids affect cell activation, proliferation, and differentiation. They modulate the levels of mediators of inflammation and immune reactions, as seen with the inhibition of interleukin-1, -2, and -6 (IL-1, -2, -6), and tumor necrosis factor synthesis (or release).20,21 Macrophage functions—including phagocytosis, antigen processing, and cell killing—are decreased by cortisol,22,23 and this decrease affects immediate and delayed hypersensitivity.
Glucocorticoids suppress monocyte and lymphocyte function (both Th1 and Th2 cells) more than polymorphonuclear leukocyte function.24 This effect is clinically important because granulomatous infectious diseases, such as tuberculosis, are prone to exacerbation and relapse during prolonged glucocorticoid therapy. The antibody-forming cells, B lymphocytes and plasma cells, are relatively resistant to the suppressive effects of glucocorticoids. Very high doses of glucocorticoids are needed to suppress antibody production.25
Pharmacokinetics
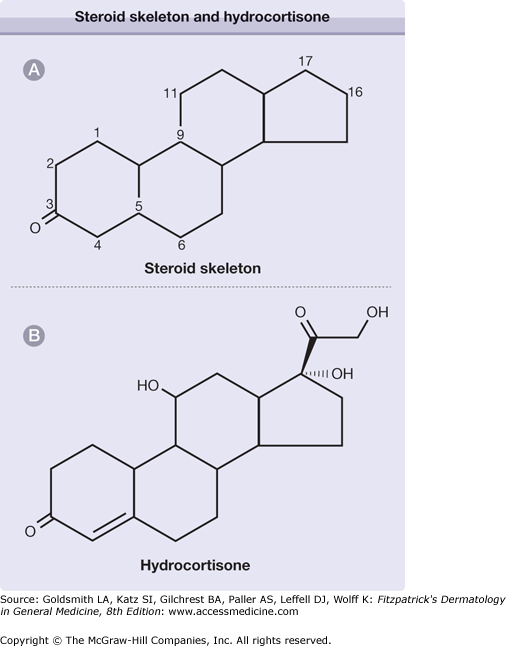
 When hydrocortisone is given in moderate-to-high doses, its mineralocorticoid effects can be deleterious, and thus synthetic analogues of cortisol have been developed that have greater anti-inflammatory properties and cause less sodium retention. Small substitutions on the basic steroid structure of three hexanes and a pentane ring (eFig. 224-0.1, eTable 224-0.1) account for the differences in plasma half-life and the relative anti-inflammatory and sodium-retaining potencies (Table 224-1). In general, most synthetic analogues bind less efficiently to CBG (about 70 percent binding). This property may explain, in part, their tendency to cause side effects at lower dosages. The 11-β-hydroxyl group in cortisol is essential for activity. Because cortisone and prednisone are 11-keto compounds, they are active only after being converted in the liver to the corresponding 11-β-hydroxyl compounds (cortisol and prednisolone) (Table 224-0.1). Patients with severe liver disease generally maintain their ability to convert the 11-keto compounds; nevertheless, some authorities suggest that only the converted active compounds should be administered to these patients.26
When hydrocortisone is given in moderate-to-high doses, its mineralocorticoid effects can be deleterious, and thus synthetic analogues of cortisol have been developed that have greater anti-inflammatory properties and cause less sodium retention. Small substitutions on the basic steroid structure of three hexanes and a pentane ring (eFig. 224-0.1, eTable 224-0.1) account for the differences in plasma half-life and the relative anti-inflammatory and sodium-retaining potencies (Table 224-1). In general, most synthetic analogues bind less efficiently to CBG (about 70 percent binding). This property may explain, in part, their tendency to cause side effects at lower dosages. The 11-β-hydroxyl group in cortisol is essential for activity. Because cortisone and prednisone are 11-keto compounds, they are active only after being converted in the liver to the corresponding 11-β-hydroxyl compounds (cortisol and prednisolone) (Table 224-0.1). Patients with severe liver disease generally maintain their ability to convert the 11-keto compounds; nevertheless, some authorities suggest that only the converted active compounds should be administered to these patients.26
Equivalent Glucocorticoid Potency (mg) | Mineralocorticoid Potency | Plasma Half-Life (min) | Duration of Action (h) | |
|---|---|---|---|---|
Short-acting | ||||
Hydrocortisone (Cortisol) | 20 | 0.8 | 90 | 8–12 |
Cortisone | 25 | 1 | 30 | 8–12 |
Intermediate-acting | ||||
Prednisone | 5 | 0.25 | 60 | 16–36 |
Prednisolone | 5 | 0.25 | 200 | 12–36 |
Methylprednisolone | 4 | 0 | 180 | 12–36 |
Triamcinolone | 4 | 0 | 300 | 12–36 |
Long-acting | ||||
Dexamethasone | 0.75 | 0 | 200 | 36–54 |
Position | |||||
|---|---|---|---|---|---|
1–2 | 6 | 9 | 11 | 16 | |
Cortisol | — | — | — | —OH | — |
Cortisone | — | — | — | =O | — |
Prednisone | Double bond | — | — | =O | — |
Prednisolone | Double bond | — | — | —OH | — |
Methylprednisolone | Double bond | CH3 | — | —OH | — |
Triamcinolone | Double bond | — | F | —OH | OH |
Dexamethasone | Double bond | — | F | —OH | CH3 |
Indications
There is a long list of indications for skin disorders (see Box 224-1). In addition, short courses of glucocorticoids may be used for a variety of forms of severe dermatitis, including contact dermatitis, atopic dermatitis, photodermatitis, exfoliative dermatitis, and erythrodermas. Low doses of glucocorticoids may be administered at bedtime if acne and hirsutism result from adrenogenital syndromes and are unresponsive to more conservative therapy. The use of glucocorticoids is controversial in the treatment of erythema nodosum, lichen planus, cutaneous T-cell lymphoma, and discoid lupus erythematosus.
|
Dosing Regimen
Systemic glucocorticoids can be administered intralesionally, orally, intramuscularly, and intravenously. The route and regimen are determined by the nature and extent of the disease being treated.
Intralesional glucocorticoid administration allows direct access to either a relatively few lesions or a particularly resistant lesion. The concentration depends on the site of injection and the nature of the lesion. Lower concentrations (2–3 mg/mL) are used on the face to prevent atrophy of the skin, whereas keloids may require concentrations of 40 mg/mL. In conditions requiring sustained effects, such as keloids and alopecia areata, longer-acting glucocorticoids, such as Aristospan, can be administered alone or mixed with the more typically used Kenalog. It is best to limit the total monthly dose of Kenalog to 20 mg to ensure that the hypothalamic-pituitary-adrenal (HPA) axis will not be suppressed.27
There are serious drawbacks to intramuscular administration because of erratic absorption and lack of daily control of the dose. Because Kenalog is longer-acting than prednisone, Kenalog has more potential side effects, including increased HPA suppression and myopathy.
When oral glucocorticoids are prescribed, prednisone is most commonly selected. Glucocorticoids are usually administered daily or every other day; although for acute disease split, daily doses can be administered. The initial dose is most often daily to control the disease process and can range from 2.5 mg to several hundred milligrams daily. If used for less than 3–4 weeks, glucocorticoid therapy can be stopped without tapering. The lowest possible dose of a short-acting agent every other morning minimizes side effects. Because cortisol levels peak at around 8 a.m., the HPA axis is least suppressed with this morning dosage, and maximal feedback suppression of adrenocorticotropic hormone (ACTH) secretion by the pituitary is already occurring. The low levels of glucocorticoids at night allow for normal secretion of ACTH. Low doses of prednisone (2.5–5 mg) at bedtime have been used to maximize adrenal suppression in cases of acne or hirsutism of adrenal origin.
Intravenous glucocorticoids are used in two situations. One is for stress coverage for patients who are acutely ill or are undergoing surgery and who have adrenal suppression from daily glucocorticoid therapy. The other is for patients with certain diseases—such as resistant pyoderma gangrenosum, severe pemphigus or bullous pemphigoid, serious SLE, or dermatomyositis—to gain rapid control of the disease and thus minimize the need for long-term, high-dose, oral steroid therapy.28 Methylprednisolone is used at a dose of 500 mg to 1 g daily because of its high potency and low sodium-retaining activity. Serious side effects associated with intravenous administration include anaphylactic reactions, seizures, arrhythmias, and sudden death. Other adverse reactions include hypotension, hypertension, hyperglycemia, electrolyte shifts, and acute psychosis. Slower administration over 2–3 hours has minimized many of the serious side effects, and as long as vital signs are determined frequently, patients without underlying renal or cardiac disease do not need to be treated in a monitored bed.29 It is important to monitor serum electrolytes before and after pulse therapy, particularly when patients are on concomitant diuretic therapy. Some studies question whether there is a role for high-dose IV pulse therapy.27
Initiating Therapy
Before therapy with glucocorticoids is begun, the benefit that can realistically be expected should be weighed against the potential side effects. Alternative or adjunctive therapies should be considered, especially if long-term treatment is contemplated. Coexisting illnesses such as diabetes, hypertension, or osteoporosis need to be considered. The predisposition of the patient to side effects should be included in an assessment of risk.
A number of considerations bear on the choice of glucocorticoids. First, a preparation with minimal mineralocorticoid effect is usually picked to decrease sodium retention. Second, the long-term oral use of prednisone or a similar drug, with an intermediate half-life and relatively weak steroid-receptor affinity, may reduce side effects. Long-term use of drugs like dexamethasone, which has a longer half-life and high glucocorticoid-receptor affinity, may produce more side effects without any better therapeutic effects. Third, if a patient does not respond to cortisone or prednisone, the substitution of the biologically active form, cortisol or prednisolone, should be considered. In general, even in severe liver disease, substitution has not proved to be very important. Fourth, methylprednisolone is used for pulse therapy because of its low sodium-retaining characteristics and high potency.
To minimize potential problems, the baseline evaluation should include a personal and family history, with special attention to predisposition to diabetes, hypertension, hyperlipidemia, glaucoma, and associated diseases that could be affected by steroid therapy. Baseline blood pressure and weight should be measured. If prolonged administration is anticipated, an eye examination and a purified protein derivative (PPD) test should be performed and an anergy panel applied. Examination for other covert infections should be based on history and physical examination. For instance, a stool culture for Strongyloides should be performed for immigrants from third-world countries and for Vietnam veterans.30 If long-term administration of glucocorticoids is anticipated, baseline spinal bone-density measurement should be obtained by quantitative computed tomography (CT), dual-photon absorptiometry, or dual-energy X-ray absorptiometry (DEXA).29
Monitoring Therapy
At follow-up visits, patients receiving chronic glucocorticoid therapy should be questioned about polyuria, polydipsia, abdominal pain, fevers, sleep disturbances, and psychological effects. There may be serious changes in effects on affect and even psychosis in patients treated with high doses of glucocorticoids. Weight and blood pressure should be monitored. Serum electrolytes, fasting blood sugar, and cholesterol and triglyceride levels should be measured on a regular basis. Stool should be examined for occult blood. Follow-up eye examinations should be performed with careful monitoring for the development of cataracts and glaucoma.
Risks and Precautions
Side Effect | Preventative Measures |
|---|---|
Hypertension | Blood pressure (baseline; repeat with each visit) |
Weight gain | Weight (baseline; repeat with each visit) |
Reactivation of infection | Purified protein derivative, anergy panel at baseline (can be done up to 12 days after starting prednisone) Hepatitis screen Consider Pneumocystis carinii pneumonia prophylaxis (Bactrim 1 DS three times a week) |
Metabolic abnormalities | Electrolytes, lipids, glucose [baseline; repeat early after starting therapy; repeat annually; more frequent monitoring with known factors (e.g., diabetes, hyperlipidemias)] |
Osteoporosis | Bone density (baseline; repeat annually is early bone prophylaxis done) Instruct about diet, exercise, other measures Calcium and vitamin D supplementation Start bisphosphonate for men, postmenopausal women Evaluate postmenopausal women for hormone replacement therapy Serum testosterone after treatment started in men; if low (<300 ng/mL), check prostate specific antigen, prostate examination before starting testosterone replacement |
Eyes | |
Cataracts Glaucoma Peptic ulceration Suppression of hypothalamic-pituitary-adrenal axis | Slit-lamp examination (every 6–12 months) Intraocular pressure examination (at 1 month and every 6 months) In patients with two or more risk factors, consider prophylaxis with and H2-antagonist or proton pump inhibitor Single, early morning doses, preferably every other day Check 8:00 am serum cortisol before tapering prednisone <3 mg/day. If <10 g/dL, repeat every 1–2 months and maintain low prednisone dose until baseline cortisol adequate |
Diet should be low in calories, fat, and sodium, and high in protein, potassium, and calcium. Protein intake is important to reduce steroid-induced nitrogen wasting.31 Use of alcohol, coffee, and nicotine should be minimized. Exercise should be encouraged.
Patients with a positive PPD or Quantiferon TB Test should be given prophylaxis with isoniazid.32 Anergic patients should have a baseline chest X-ray to search for evidence of previous tuberculosis. Fevers or focal findings should be evaluated with appropriate cultures and diagnostic approaches. Some advocate use of Bactrim prophylaxis (1 DS Bactrim 3 days a week) against Pneumocystic carinii when patients receive concomitant cytotoxic therapy.26
Immunization with live vaccines can be done if the duration of glucocorticoid use is less than 2 weeks at any dose, if the dose of glucocorticoid is <2 mg/kg or 20 mg/day of any duration, and if long-term alternate-day treatment with short-acting preparations is done. Immunization with live vaccines should not be done for at least 3 months after receiving high doses of glucocorticoids (>2 mg/kg or greater than 20 mg/day) for more than 2 weeks.33
Although there is controversy about whether an increase in the incidence of peptic ulcer disease occurs in otherwise unaffected patients receiving glucocorticoids, there is almost a ninefold increase in patients taking both glucocorticoids and nonsteroidal anti-inflammatory agents.32,34,35 In patients with two or more risk factors (such as those taking nonsteroidal anti-inflammatory medications, a previous history of peptic ulceration, advanced malignant disease, or a total dose of glucocorticoids >1,000 mg), prophylaxis may be considered. Prophylaxis can include antacids, H2-receptor blockers (cimetidine, ranitidine, nizatidine, or famotidine with the evening meal), or proton pump inhibitors (Prilosec or Prevacid).
Patients receiving daily glucocorticoid therapy for longer than 3–4 weeks must be assumed to have adrenal suppression that requires tapering of the glucocorticoids to allow for recovery of the HPA axis. Tapering is best performed by switching from a single daily dose to alternate-day doses, followed by a gradual reduction of the amount of the drug. The daily dose is first gradually tapered to 40 or 50 mg of prednisone. Then either the dose can be kept constant on one day and reduced on the alternate day by 5-mg decrements down to 5 mg/day, or, the steroid dose can be increased on one day and reduced by a similar amount on the alternate day. More studies are needed to determine optimal tapering schedules.36
After the prednisone dose is tapered to 5 mg on alternate days, the need for maintenance therapy must be assessed. The 8:00 am plasma cortisol level is measured 4 weeks after the 5-mg dose has been reached. The morning dose of prednisone is held until the plasma cortisol level is determined. If the plasma cortisol level is less than 10 μg/dL, the alternate-day prednisone dose should be decreased by 1 mg every 1–2 weeks to a maintenance dose of 2 mg/day. Then the 8:00 am plasma cortisol level should be rechecked every 2 months until it is greater than 10 μg/dL, at which point maintenance glucocorticoids can be terminated.37 Recovery of the HPA axis can take longer than 9 months.38
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


