Squamous Cell Carcinoma: Introduction
|
Cutaneous squamous cell carcinomas (SCCs) are malignant neoplasms derived from suprabasal epidermal keratinocytes. These and basal cell cancers are the nonmelanoma skin cancers that represent the most common malignancies in humans. Whereas basal cell carcinoma (BCC) (see Chapter 115) is thought to arise de novo, SCC probably evolves in most cases from precursor lesions of actinic keratosis (AK) and Bowen disease (SCC in situ) (see Chapter 113). This chapter focuses on clinical aspects of invasive SCC. Cutaneous SCC represents a broad spectrum of disease ranging from easily managed, superficially invasive cancers to highly infiltrative, metastasizing tumors that can result in death. The clinical presentation can be variable despite the existence of easily identified typical lesions. The cellular and molecular aspects of SCC carcinogenesis are discussed elsewhere (see Chapter 113).
Historical Aspects
The precise incidence of BCC and SCC is unknown, because these cutaneous malignancies are not generally documented by the National Cancer Institute or most state cancer registries. However, it is generally accepted that well over 1 million cases are diagnosed in the United States each year, with approximately 200,000 representing SCC.10 Although less common than BCC, SCC carries a risk of metastasis and thus accounts for the majority of the several thousand deaths attributable to nonmelanoma skin cancer each year. By comparison, cutaneous melanoma accounts for only 60,000 cases, but approximately 9,000 deaths, annually.11 Similar trends for SCC have been noted in Australia12 and the Caribbean.13
SCC is strongly associated with advanced age, and a sharp increase in incidence is seen after age 40 years.14 Today, the lifetime risk of SCC among whites is approximately 15%, almost double that of two decades ago. Increased exposures to ultraviolet (UV) radiation (through greater use of tanning salons, increased time spent outdoors, changes in clothing styles, and ozone depletion) and greater longevity have been suggested as possible causes for the increase in disease. It is likely that this trend will continue as a result of further depletion of the ozone layer and the aging of the US population. The rising incidence of SCC over the past several decades has been paralleled by a 20% decrease in mortality, attributed largely to increased public awareness and aggressive treatment of high-risk lesions.15 After a diagnosis of SCC, patients have a 44%–50% cumulative risk of developing another nonmelanoma skin cancer (18%–30% risk of SCC) in the subsequent 3–5 years.16 In addition, these patients are at increased risk for extracutaneous–cancers.17
Squamous cell cancer is twice as common in men as in women, probably as a result of greater lifetime UV exposure in men. Similarly, longer hairstyles and use of lipstick may account for the lower frequency of SCC on the ears and lips of women. There is an inverse relationship between skin pigmentation and SCC incidence, largely because of the protective effect of eumelanin. Thus, persons with white skin, blue eyes, fair complexion, red hair, and Celtic ancestry who tan poorly are at greatest risk. Increased pigmentation is associated not only with a lower incidence of SCC, but also with an inversion in the BCC/SCC ratio. In comparison with whites, the incidence of SCC is decreased by 30-fold in American blacks and the BCC/SCC ratio falls to 0.8:1.14 Tanzanian albinos all develop SCC by young adulthood, and the ratio of BCC to SCC is only 0.2:1. Asians and Polynesians, with intermediate skin pigmentation, have correspondingly intermediate levels of SCC. A gene (MC1R) involved in melanogenesis that encodes the melanocortin 1 receptor is a major determinant of skin pigmentation and hair color. The MC1R gene is highly polymorphic, with more than 20 variants described.18 Several variant MC1R alleles are associated with increased risk of SCC that is independent of skin type and hair color.
Etiology and Pathogenesis
There are a number of factors, including both acquired and genetic skin conditions that may predispose to SCC (Table 114-1). Patients often demonstrate a multiplicity of factors that together are sufficient to induce SCC development. For example, a given skin site may be exposed to both UV radiation and another environmental carcinogen.
|
Most SCCs develop from precursor lesions such as AKs or Bowen disease (see Chapter 113).
UV radiation is considered the predominant risk factor for SCC. Importantly, there is a linear correlation between the incidence of SCC and exposure to UV radiation. The incidence of SCC has been reported to double with each 8°–10° decline in geographic latitude and is highest at the equator.19 World War II veterans stationed in the Pacific developed much higher rates of SCC than did their colleagues who served in Europe.20 Similarly, SCC is more prevalent in Japanese people who emigrated to Hawaii than in those who remained in Japan.21 Excessive UV radiation appears to be related more to the development of SCC than to the development of BCC. Rates of SCC rise more rapidly than those of BCC with increasing UV exposure,22 and UV radiation-induced skin cancers in mice are almost exclusively SCCs rather than BCCs.23 Moreover, in patients receiving long-term therapy with psoralen plus ultraviolet A (UVA) radiation for treatment of psoriasis there is an associated 30-fold increase in nonmelanoma skin cancers, most of which are SCCs.24
There is a strong association between SCC and exposure to ionizing radiation. In one survey of SCC patients, an association with radiation therapy was observed only in those whose skin was likely to sunburn (see Chapter 113).
Numerous occupational and environmental carcinogens, such as arsenic and aromatic hydrocarbons, predispose to the development of SCCs. With the exception of 3-methylcholanthrene and anthramine, chemical carcinogens generally produce SCCs rather than BCCs.25 Exposures to insecticides and herbicides have also been associated with SCCs. In addition, smoking and alcohol use are strongly associated with SCCs of the oral cavity.
Chronic immunosuppression may lead to an increase in SCCs, primarily on sun-exposed sites.26 An 18-fold increase in SCC has been reported in renal transplant patients27; these tend to appear 3–7 years after the onset of long-term immunosuppressive therapy, with corticosteroids, azathioprine, and cyclosporine most frequently implicated. With the increase in the total number of organ transplant patients, management of SCCs in this population is becoming more important. In patients with leukemia and lymphoma, SCCs are both increased and more aggressive.28 Although multiple SCCs have been described in patients infected with human immunodeficiency virus, advanced human immunodeficiency virus infection has generally not been associated with an increased incidence of SCC, possibly because many patients do not live long enough to develop them.
Historically, SCC was associated with both burn scars and chronic ulcers as noted earlier, but such associations are seldom seen today. Also rare but reported is the development of SCCs in the context of chronic infections, particularly those associated with draining sinuses and scarring, such as perianal pyoderma, osteomyelitis, chromomycosis, hyalohyphomycosis, granuloma inguinale, lupus vulgaris, and leprosy. Chronic inflammatory processes, particularly those associated with scarring, such as venous ulcer, snakebite ulcer, discoid lupus erythematosus, oral lichen planus, morphea, lichen sclerosus, pilonidal cyst, acne conglobata, hidradenitis suppurativa, Hailey–Hailey disease, dissecting folliculitis of the scalp, and necrobiosis lipoidica, all can give rise to SCCs. An exception is vaccination scars, which are associated with BCCs rather than SCCs. SCCs have also been observed in transplanted skin, epidermal cyst, dental cyst, and dermoid cyst.
Long-term heat exposure can lead to SCCs. The role of thermal radiation in the development of skin cancer has long been recognized in many cultures, where common practices include placing hot ashes under the clothes to keep warm in winter or smoking opium while lying on heated beds. The incidence of SCCs is increased in persons who habitually sit in front of heating stoves and at sites of erythema ab igne (see Chapter 113).
A role for human papillomavirus (HPV) infection has been well established in some types of SCCs. Verrucous carcinoma appears to be associated with several HPV types, as noted later. Head and neck and periungual SCCs are frequently associated with HPV-16. Patients with epidermodysplasia verruciformis are chronically infected with HPV, most commonly type 5, and one-third of these patients ultimately develop SCCs (see Chapters 113 and 196). Recently, the MCPyV polyoma virus, originally discovered in Merkel cell carcinoma, was identified in approximately 15% of cutaneous SCCs from immunocompetent patients.A An etiological role for MCPyV in SCC remains to be demonstrated.
A variety of heritable diseases predispose to SCC development. Patients with oculocutaneous albinism develop predominantly SCCs (rather than BCCs) at an early age (see Chapter 73). Xeroderma pigmentosum (see Chapters 110 and 139), a disorder of DNA repair, is also characterized by early development of SCCs. SCCs have been reported to develop in the Mibelli, disseminated superficial actinic, and palmaris et plantaris disseminata forms of porokeratosis (see Chapter 52), and in oral lesions of dyskeratosis congenita. As noted in Section “Viral Infection,” lesions of epidermodysplasia verruciformis can degenerate into SCCs (see Chapter 196). Finally, patients with the dystrophic form of epidermolysis bullosa are at increased risk for SCC (see Chapter 62).
As in most cancers, the development of SCC from normal keratinocytes begins with mutations in the cellular DNA and genomic instability. Alterations in gene expression lead to loss of growth controls, penetration of the basement membrane, and ultimately invasion into surrounding tissue. Along the pathway to SCC, keratinocytes become resistant to apoptosis (programed cell death) and immune attack.
Most analyses of genetic alterations in SCCs have been performed in cases of oral or head and neck SCCs. Chromosomal deletions (loss of heterozygosity) commonly involve chromosomes 3, 9, 11, and 17; the regions most commonly identified include 9p21 and 17p13 where the INK4A (p16/Arf) and p53 tumor suppressors, respectively, are located.29 Similar genetic lesions were found in a study of young patients (younger than 40 years of age).30 It is unclear whether these genetic markers will serve as useful prognostic indicators.
A role for p53, cyclin D1, human telomerase reverse transcriptase, p16, and thrombospondin 1 has been identified in the multistep process of human skin carcinogenesis.31
Apoptosis of keratinocytes that have sustained UV radiation-induced DNA damage, termed sunburn cells, requires the p53 tumor suppressor and represents a key protective mechanism against skin cancer by removing premalignant cells that have acquired mutations. In keratinocytes, UV radiation upregulates p53,32 which delays cell cycle progression until DNA damage can be repaired or facilitates cell elimination by apoptosis.33 Compromise of p53 function could undermine this apoptosis-based defense mechanism, giving UV-damaged cells a selective advantage to survive additional cycles of UV exposure.34 Further impairment of p53 and other genes through additional UV radiation-induced mutations may then lead to even greater resistance to apoptosis, increased proliferation, and ultimately development of SCC. The increased susceptibility of p53-deficient mice to UV radiation-induced SCC35 highlights this protective role of p53.
Consistent with the scenario described in Section “p53 in the Defense Against Skin Cancer,” mutations in the p53 gene are a common finding in SCC.36 In most cases, these are C→T single base and CC→TT tandem transition mutations at dipyrimidine sequences, i.e., “UVB-signature” mutations.37 Most SCCs exhibit loss of heterozygosity with respect to p53 and isolated mutations on the remaining allele. In one study, the p53-apoptosis pathway was disrupted in 50% of oral SCCs. With respect to SCC precursors, p53 mutations were found in up to 75% of AK and SCC in situ lesions.38 Interestingly, although different p53 mutations were found in separate AKs, all cells within a single precursor lesion had the same mutation.31 Mutations in p53 can also be detected in keratinocytes from clinically normal sun-exposed skin.39 Keratinocytes with p53 mutations occur in clonal patches that are larger and more frequently in sun-exposed skin.40 These findings substantiate a clonal basis for UV radiation-induced SCC and suggest that p53 mutation is an early event in the development of SCC.
In addition to undergoing mutation, p53 can be compromised in keratinocytes infected with HPV. The E6 protein encoded by oncogenic HPV types binds p53 and targets it for rapid degradation, which disables the p53-apoptosis pathway. This is a primary mechanism by which HPV infection predisposes to SCC (see Chapter 196).
In addition to dysregulation of p53, dysregulation of other apoptotic regulatory proteins has been described in SCC. In a study of vulvar SCC, expression of the apoptotic inhibitor Bcl-2 correlated with metastasis.41 Similarly, in esophageal SCC, expression of the apoptotic inhibitor Bcl-XL correlated with tumor invasion and metastasis.42 In SCC of the tongue, low apoptotic index and decreased expression of the proapoptotic Bcl-2-associated X protein (Bax) correlated significantly with poor prognosis, whereas low Bcl-2 expression was associated with a favorable clinical outcome.43 Expression of the antiapoptotic Bcl-2-associated athanogene 1 (BAG-1) was associated with nodal metastasis in oral SCC.44 Consistent with these observations, transgenic mice expressing Bcl-2 and Bcl-XL in the skin exhibit increased susceptibility to chemical-induced tumorigenesis. In addition to these Bcl-2 family members, the inhibitor of apoptosis protein survivin is expressed in both SCC and precursor lesions,45 and in one study its expression correlated with aggressive tumor phenotype.46 Interestingly, survivin may be negatively regulated by the binding of p53 to its promoter.47 More recent studies in transgenic mice have yielded paradoxical results, which suggests that apoptosis may be required in the initial phase of UV radiation-induced clonal expansion.48
Working with UV radiation-induced SCCs in mice, Kripke and colleagues first demonstrated the importance of immunosuppression in UV radiation-induced SCC in the 1970s (see Chapter 90). They found that although UV radiation-induced SCC was promptly rejected when transplanted into genetically identical recipient mice, the transplanted tumors grew rapidly, and rejection did not occur if recipient mice were first treated with a subcarcinogenic dose of UV radiation. These experiments suggested that UV radiation not only induced SCC but also impaired the ability of host animals to mount protective immune responses against foreign tumor antigens.
In white men and women, the majority of SCCs arise on sun-exposed areas such as the head, neck, and dorsal hands. SCC of the legs is more common in women.49 On the other hand, in blacks SCCs tend to be distributed equally on sun-protected and sun-exposed areas.50 SCC typically presents in solitary fashion, arising from precursor lesions as noted earlier. An exception is in immunosuppressed patients, who may manifest eruptive SCCs.
(See Chapter 113)
AKs often occur as a multiplicity of lesions, ranging in size from pinpoint to over 2 cm, and the borders are usually ill defined. A dry adherent scale gives them a rough, gritty texture. By contrast, lesions of Bowen disease are usually solitary, sharply demarcated, scaling papules or plaques, often initially mistaken for eczema, psoriasis, or lichen simplex. The latter disorders are often pruritic, whereas Bowen disease is usually not. In sun-protected sites, Bowen disease may have a noneczematous appearance. For example, it may appear verrucous in the anogenital area, nail bed, and eyelid, and as a dark patch or oozing erythematous plaque in intertriginous areas. These precursor lesions are usually asymptomatic, and the development of tenderness, induration, erosion, increased scale, or enlarging diameter may herald evolution into SCC. Typically, a patient with multiple AKs may present with a single lesion that gradually becomes more prominent than the rest (Fig. 114-1), or with a solitary, persistent, nonpruritic, scaling patch that is unresponsive to treatment with topical steroids.
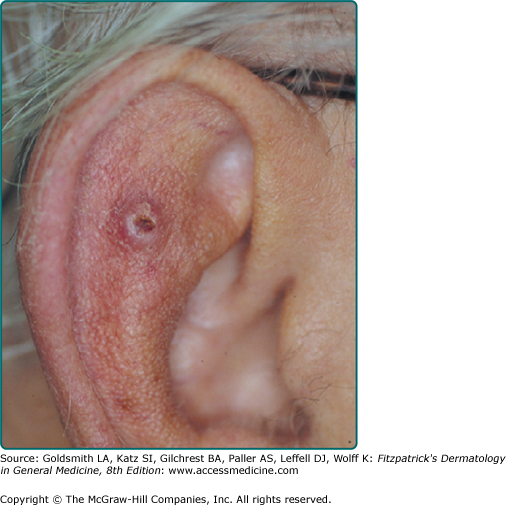
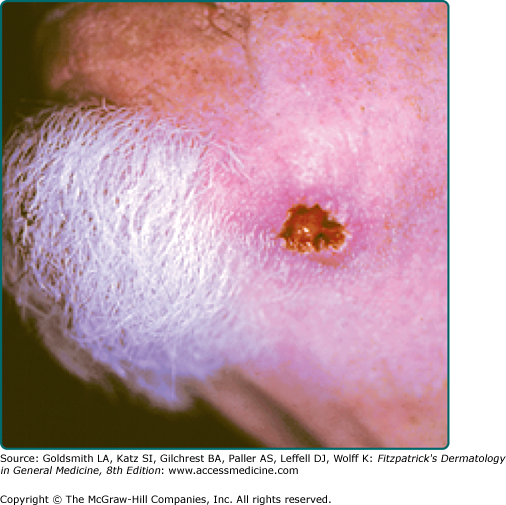
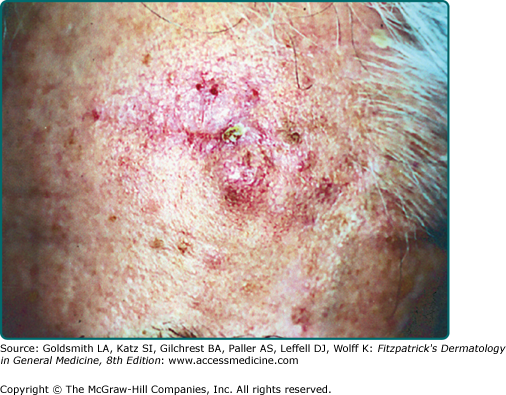
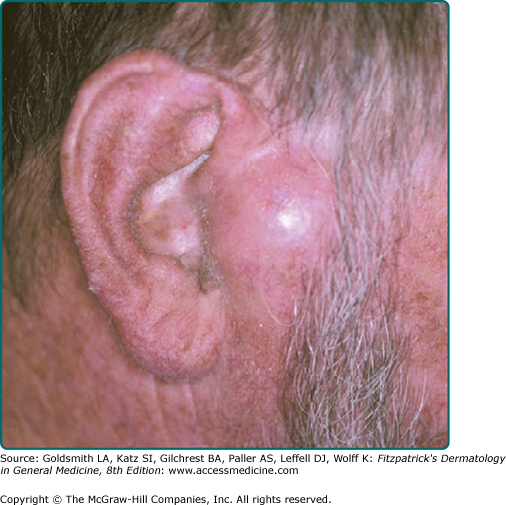
A firm, flesh-colored or erythematous, keratotic papule or plaque is most common (see Fig. 114-1), but SCCs may also be pigmented. Other presentations include as an ulcer (Fig. 114-2), a smooth nodule (Fig. 114-3), or a thick cutaneous horn. SCC may also be verrucous or present as an abscess, particularly if in a periungual location (see Fig. 113-9 in Chapter 113). The margins may be indistinct. With enlargement, there is usually increased firmness and elevation. Progressive tumor invasion ultimately results in fixation to underlying tissues. Especially in the head and neck region, an enlarged lymph node nearby that is firm and nontender may indicate tumor metastasis (Fig. 114-4).
SCC of the oral cavity usually occurs in patients with a long history of cigarette smoking, tobacco chewing, or alcohol use, but it has now been documented in younger adults without these traditional risk factors. There is a male predominance, and the palate and tongue are the most common sites. Oral SCC most commonly evolves from lesions of erythroplakia and is usually asymptomatic (see Chapter 113). Distinct patterns include a persistent rough red patch or granular velvety red plaque that ultimately becomes firm and nodular. Surprisingly, the risk of transformation to SCC does not appear to correlate with the degree of epithelial dysplasia.51 The floor of the mouth, ventrolateral tongue, and soft palate are considered high-risk sites. It may also present as a peritonsillar abscess.
SCC of the lower lip begins as a roughened papule of actinic cheilitis or scaly leukoplakia, with slow progression to a tumor nodule (Fig. 114-5). Clinical clues associated with evolving SCC include persistent lip chapping with localized scale or crust, red and white blotchy atrophic vermilion zone of the lip, indistinct or “wandering” vermilion border, and small fissuring or ulceration within an area of indurated actinic cheilitis. Symptoms of underlying pain or altered sensation should be investigated as a potential sign of perineural invasion.
Figure 114-5
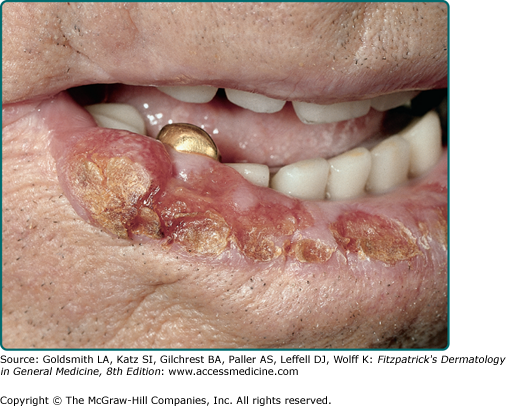
Squamous cell carcinoma of the lower lip that developed in the setting of habitual sun exposure and actinic cheilitis. There is a large but subtle nodule, better felt than seen, on the lower lip. There are areas of hyperkeratosis and ulceration. Metastasis to draining lymph nodes can occur.
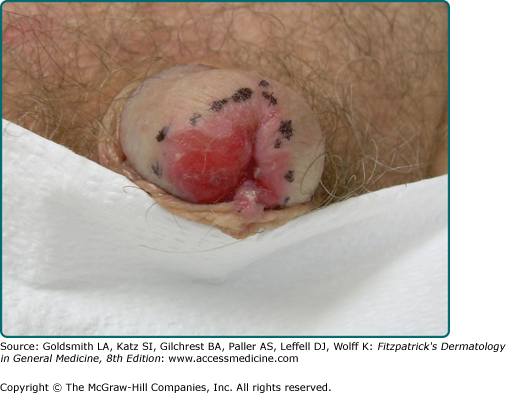
SCC of the vulva most commonly occurs on the anterior labia majora, beginning as a small warty nodule or an erosive erythematosus plaque. These lesions may be asymptomatic but more often are associated with pruritus or bleeding. Lesions of lichen sclerosus are another common precursor of SCC of the vulva. SCC of the cervix is associated with HPV infection, most commonly with type 16. SCC of the scrotum begins as a small pruritic verrucous lesion that becomes friable with increasing size. SCC of the penis usually occurs in uncircumcised males (see eFig. 114-5.1) and, although very uncommon in Western countries, may account for 10% of cancers in places where genital hygiene is poor. A distinct precursor of penile SCC is erythroplasia of Queyrat (see Chapter 113), characterized by a velvety red plaque. In addition to lack of circumcision, penile SCC has been associated with a history of condyloma and phimosis and lichen sclerosus et atrophicus (see Chapters 65 and 78). The genitalia were once thought to be a common location for SCC after long-term therapy with psoralen and UVA radiation, but this complication can be avoided by shielding the genitalia during treatment, and such an association is rarely seen today. Perianal SCC may also occur (Fig. 114-6).
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


