Key Terms
Scleroderma
Progressive systemic sclerosis
Scleredema
Scleredema adultorum
Scleredema of Buschke
Sclerosing and fibrosing disorders manifest as a hardening or induration of the skin due to the overproduction (fibrosis) and/or thickening (sclerosis) of collagen bundles. Sometimes, organ systems beyond the skin may be affected. Causes include collagen vascular disease and drugs, the latter taken orally or injected.
- •
Localized scleroderma (morphea)
- •
Limited scleroderma (CREST syndrome)
- •
Systemic scleroderma
- •
Drug-induced sclerosis (e.g., bleomycin)
- •
Eosinophilic fasciitis
- •
Graft-versus-host disease
- •
Mixed connective tissue disease
- •
Morphea
- •
Nephrogenic systemic fibrosis
- •
Porphyria cutanea tarda
- •
Progressive systemic sclerosis
- •
Scleredema
- •
Scleromyxedema
Important History Questions
Do you have a history of cold intolerance in your fingers or known Raynaud phenomenon?
This is an important question because systemic scleroderma, limited scleroderma (formerly CREST syndrome [ c alcinosis, R aynaud phenomenon, e sophageal dysmotility, s clerodactyly, and t elangiectasia]), and mixed connective tissue disease (MCTD) can present first in the skin, and the presence of Raynaud phenomenon may be an indication to pursue additional investigation.
Have you noticed swelling in your hands?
Swelling or edema of the hands—puffy hands—can be an early sign of evolving limited or systemic scleroderma or MCTD.
Do you have any difficulty in swallowing solid foods?
Dysphagia can be seen in those with systemic or limited sclerosis or MCTD.
What medications do you take?
This is an important question because some disorders, such as drug-induced sclerosis, eosinophilic fasciitis, and Texier disease, can be drug induced. Texier disease is not discussed in this chapter, but it produces a morphea-like or localized fasciitis-like syndrome at the site of vitamin K injection.
Have you had any recent infections?
Some cases of eosinophilic fasciitis occur after a recent streptococcal or viral infection.
Do you have any other medical conditions?
Other medical conditions of interest include diabetes and multiple myeloma (associated with scleredema). A history of bone marrow transplantation could suggest sclerodermatous graft-versus-host disease.
Have you had any gadolinium-enhanced magnetic resonance imaging (MRI) studies?
Most patients have gadolinium-enhanced MRI studies without consequence, but nephrogenic systemic fibrosis can occur in patients administered these agents in the presence of significant renal disease.
Important Physical Findings
What is the distribution of the fibrosis?
Clinically observed fibrosis or sclerosis can be localized (morphea), confined to limited areas (CREST syndrome, eosinophilic fasciitis), or generalized (systemic scleroderma, generalized morphea, MCTD, or nephrogenic systemic fibrosis).
Are there any other types of skin lesions?
Matlike telangiectasias are often seen on the oral mucosa, face, neck, upper chest, and palms in limited scleroderma (CREST syndrome) but may occur also in systemic scleroderma and MCTD. Also, periungual telangiectasias, often best visualized with an ophthalmoscope, are a feature of limited and systemic scleroderma and MCTD.
Are there any changes in skin color?
Hyperpigmentation of the skin is a feature observed in systemic scleroderma, localized scleroderma (morphea), and eosinophilic fasciitis.
Morphea
ICD10 code L94.0
COLLAGEN VASCULAR DISEASE
Pathogenesis
Morphea is a localized abnormality of thickened collagen bundles in the dermis and sometimes in the subcutis. In Europe and Japan, Borrelia burgdorferi DNA has been detected in some biopsies of morphea, suggesting a possible role in pathogenesis, but this has been disputed, and it is not a feature of most cases in the United States. Importantly, morphea is a cutaneous-limited form of collagen vascular disease. Although some experts have promoted the term localized scleroderma, many dermatologists prefer retention of the older term to distinguish it from localized and systemic scleroderma, in which consequences beyond the skin are realized. About one-third of patients with morphea are positive for antinuclear antibody (ANA), usually in low titer. Morphea can also occur in the radiation field after treatment of breast cancer.
Clinical Features
- •
Morphea is more common in women than men (M : F = 3 : 1) and, although it can occur at any age, it is most common in children, adolescents, and young adults.
- •
The primary early lesion of morphea is an erythematous to violaceous plaque of variable size, but, in some patients, the inflammatory stage is not observed.
- •
The trunk and proximal extremities are affected most often.
- •
Plaque size varies from millimeters to larger than 20 cm.
- •
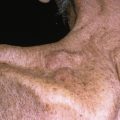
As lesions of morphea progress, the center becomes white or yellow-white and indurated, with a violaceous peripheral edge (so-called “lilac ring”; Fig. 21.1 ).
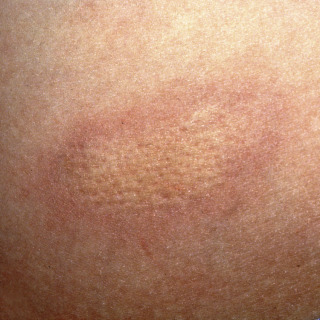
Fig. 21.1
Patient with early lesion of morphea demonstrating central area of thickened collagen and a lilac rim of discoloration.
(From the Fitzsimons Army Medical Center Collection, Aurora, CO.)
- •
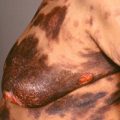
Mature lesions lose the lilac ring and, instead, manifest with discoloration, ranging from tan to brown ( Figs. 21.2 and 21.3 ) to white ( Fig. 21.4 ) or even yellow-white.
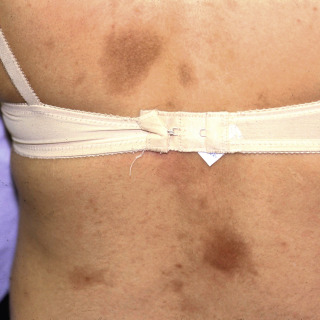
Fig. 21.2
Patient with multiple brownish lesions of morphea on the back. The lesion on the right lower back is brownish-red, indicating an actively inflamed lesion.
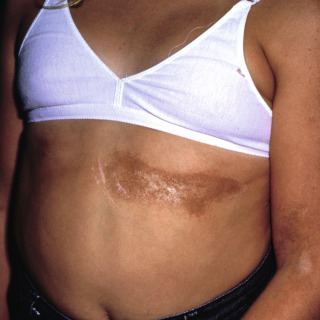
Fig. 21.3
Patient with long-standing brownish indurated plaque that has retracted. The white areas represent lichen sclerosis et atrophicus (LS&A)-like areas.
(From the Fitzsimons Army Medical Center Collection, Aurora, CO.)
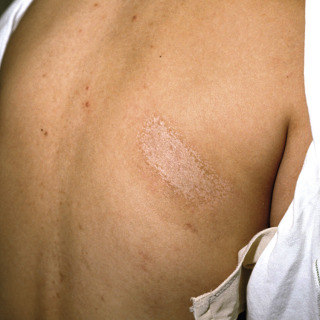
Fig. 21.4
Patient with long-standing morphea with hypopigmented surface reflecting lichen sclerosis et atrophicus (LS&A)-like changes.
- •
Mature lesions may demonstrate depression in relation to the surrounding skin (see Fig. 21.3 ).
- •
Most patients are asymptomatic, although mild pruritus may be present.
- •
Clinical variants include the following:
- –
Guttate morphea—multiple to numerous small lesions
- –
Linear morphea—linear plaques, often across joint spaces, which may hinder movement
- –
Hemifacial atrophy (coup de sabre) —a linear form that affects the heads of children or adolescents, yielding facial asymmetry and scarring hair loss
- –
Bullous morphea—a rare variant characterized by occasional intermittent blisters
- –
Morphea with lichen sclerosus–like changes—the indurated findings of morphea, but with an overlying porcelain white or slightly blue-white color
- –
Generalized morphea—involves most of the skin
- –
Diagnosis
- •
The diagnosis of morphea can usually be made on clinical grounds, with a classic indurated oval or linear plaque, but if an early lesion has a lilac ring, the diagnosis is even more certain.
- •
Unusual presentations or atypical variants may require a punch biopsy (typically, 4, 5 or 6, mm) or incisional biopsy that includes fat. A shave biopsy is inappropriate to establish the diagnosis of morphea.
Treatment
- •
No treatment is an option for limited and asymptomatic disease because most cases spontaneously resolve.
- •
Both psoralen with UVA light (PUVA) or UVA1 phototherapy have limited availability but are probably the treatments of choice.
- •
If phototherapy is not an option, potent topical corticosteroids, such as clobetasol, or topical calcipotriene 0.005% applied bid, may yield benefit.
- •
Patients with linear morphea that crosses a joint space should be referred for physical therapy.
Clinical Course
Most patients demonstrate gradual improvement, and even resolution, over a period of 3 to 5 years. Atrophy may not resolve in all cases, even if the inflammation and progression are halted. Patients with localized variants of morphea are not more likely to develop systemic scleroderma.
 Scleroderma
Scleroderma
ICD10 code M34.0
COLLAGEN VASCULAR DISEASE
Pathogenesis
Systemic scleroderma, also known as progressive systemic sclerosis, the most severe form of scleroderma, is an autoimmune collagen vascular disease that is characterized by thickening of collagen bundles. As the name implies, systemic scleroderma causes vascular changes in multiple organ systems. Numerous autoantibodies have been identified in systemic scleroderma, including antinuclear antibodies (anti–topoisomerase I, anti–RNA polymerase I and III), anti–fibrillin I, and autoantibodies directed against endothelial cells. The pathogenesis of systemic scleroderma remains poorly understood.
Clinical Features
- •
Systemic scleroderma is more common in women than men (M : F = 3 : 1).
- •
Raynaud phenomenon is a frequent early manifestation (~70% of patients).
- •
In early disease, there may be edema of the hands, which can progress into so-called bound-down sclerosis.
- •
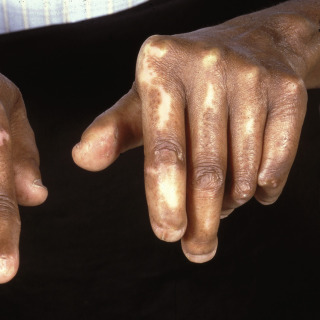
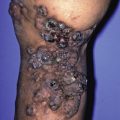
In systemic scleroderma, sclerotic changes eventually involve most of the skin and may produce so-called stovepipe extremities, with shiny, taut skin ( Fig. 21.5 ) that can impair joint mobility.
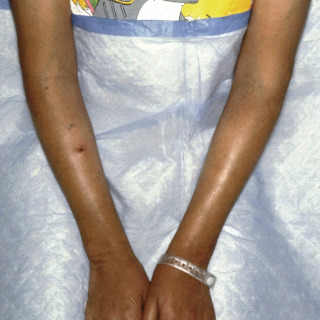
Fig. 21.5
Progressive systemic sclerosis in a young lady demonstrating fibrotic tightly bound-down shiny skin of the arms.
(From the Fitzsimons Army Medical Center Collection, Aurora, CO.)
- •
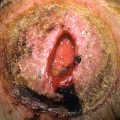
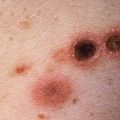
As the disease progresses, other cutaneous manifestations include periungual telangiectasias, mat-like telangiectasias of the oral mucosa or skin, hyperpigmentation or hypopigmentation ( Fig. 21.6 ), nail dystrophy, digital infarcts ( Fig. 21.7 ), and calcinosis cutis ( Fig. 21.8 ).