Purpuric and hemorrhagic (P&H) disorders may be caused by systemic illness, potentially fatal infections (e.g. meningococcemia), coagulopathies, vascular occlusive disorders (e.g. cryoglobulinemia), and inflammatory disorders of blood vessels (e.g. vasculitis).
- •
Acute bacterial endocarditis
- •
Acute hemorrhagic edema
- •
Cryofibrinogenemia
- •
Cryoglobulinemia
- •
Disseminated candidiasis
- •
Disseminated gonococcal infection
- •
Disseminated staphylococcal infection
- •
Henoch-Schönlein purpura
- •
Meningococcemia
- •
Pityriasis lichenoides et varioliformis acuta
- •
Progressive pigmented purpura
- •
Purpura fulminans
- •
Rocky Mountain spotted fever
- •
Solar purpura
- •
Traumatic purpura
Important History Questions
How long have the lesions been present?
Some P&H disorders occur acutely, such as Rocky Mountain spotted fever, whereas other conditions present with a chronic pattern (e.g., progressive pigmented purpura).
Have you had more than one episode?
Some P&H disorders present as a singular event (e.g., meningococcemia, disseminated gonococcal infection), whereas others present as multiple episodes (e.g., pernio, cryofibrinogenemia, Henoch-Schönlein purpura).
What medications do you take?
Some P&H disorders, such as leukocytoclastic (e.g., hypersensitivity) vasculitis and progressive pigmented purpura, may be induced by drugs.
Do you have other known medical conditions?
Collagen vascular disease, in particular, may be associated with leukocytoclastic (hypersensitivity) vasculitis, pernio and chilblains, and cryofibrinogenemia.
Do you or any member of your family have a history of blood clots or a clotting disorder?
Purpura fulminans occurs more often in people with clotting disorders, which can be familial.
Is there a recent history of tick bites or exposure to ticks, especially to a dog with ticks?
This is an important question for establishing a presumptive diagnosis of Rocky Mountain spotted fever.
Important Physical Findings
What is the distribution of the lesions?
This can be an important clue to the diagnosis because some conditions characteristically involve certain anatomic sites. For example, pernio is usually confined to the toes, whereas the rash of Rocky Mountain spotted fever usually begins in acral locations. Some disorders, such as cryoglobulinemia and cryofibrinogenemia, are located at sites exposed to the cold.
Is there any physical evidence of active arthritis or arthralgias?
Some diseases, such as Henoch-Schönlein purpura, cryoglobulinemia, and disseminated gonococcal infection, are often associated with arthritis or arthralgias.
Are there other skin lesions present?
The size and characteristics of the cutaneous hemorrhage may provide clues to the underlying cause. Small nonpalpable purpura (≤4 mm) is usually a noninflammatory event and it is most common in the setting of thrombocytopenia (platelet count <20,000/mm 3 ) or abnormalities of clotting factors. Small vessel vasculitis usually produces palpable purpura, with elevated papules of up to 10 mm that are surrounded by a rim of erythema, even in addition to the purpura. Large areas of nonpalpable purpura on photo-exposed skin is associated with acquired structural anomalies due to sun damage (e.g., solar purpura). Purpura with a retiform (net-like) pattern is more often associated with vascular occlusion (e.g., calciphylaxis).
Henoch-Schönlein Purpura
ICD10 code D69.0
INTERNAL ETIOLOGY
Pathogenesis
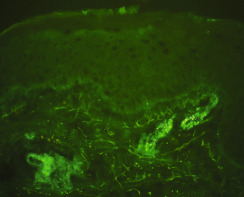
Henoch-Schönlein purpura (HSP) is a variant of leukocytoclastic vasculitis (LCV) that affects mostly children, but can also affect adults. HSP is a small vessel vasculitis characterized by circulating immunoglobulin A (IgA)-mediated immune complexes that are deposited in vessel walls ( Fig. 20.1 ). This leads to inflammation and vessel damage. Precipitating factors include viral infections (e.g., parvovirus B19, hepatitis B, hepatitis C, human immunodeficiency virus) and bacterial infections ( Streptococcus, Salmonella, and Shigella spp. and Staphylococcus aureus ). Renal disease is a feared consequence of HSP, but it occurs in a minority of patients.
Clinical Features
- •
HSP usually affects children between 2 and 10 years of age, but it may also affect younger children, adolescents, and adults.
- •
HSP is characterized by the rather abrupt onset of symmetrically distributed palpable purpura ( Figs. 20.2 and 20.3 ).
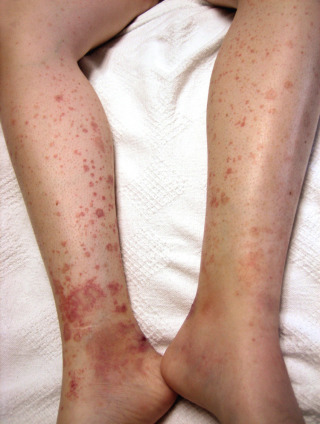
Fig. 20.2
Acute Henoch-Schönlein purpura in a child.
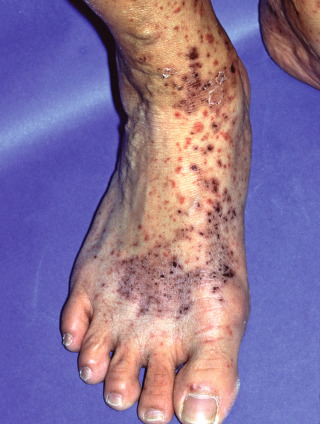
Fig. 20.3
Developed Henoch-Schönlein purpura in a young adult.
- •
In about 5% of cases, there may be pustular lesions ( Fig. 20.4 ), hemorrhagic bullae, or ulcerations.
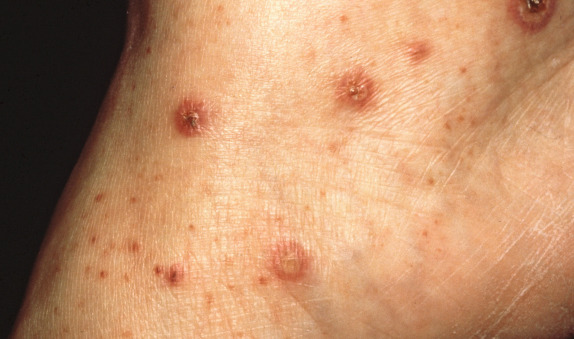
Fig. 20.4
Close-up of a patient with Henoch-Schönlein purpura illustrating classic palpable purpura, a pustular lesion, and older lesions demonstrating early crusting.
(From the Fitzsimons Army Medical Center Collection, Aurora, CO.)
- •
The legs are most often affected, followed by the buttocks.
- •
Young children usually demonstrate urticarial lesions, facial edema, and/or scrotal swelling.
- •
Systemic features include arthritis and arthralgia, headache, nephritis, and gastrointestinal (GI) symptoms, including GI angina and bowel infarction.
Diagnosis
- •
The occurrence of palpable purpura in a younger child, particularly in association with arthritis and arthralgias, headache, and GI upset, should suggest HSP. Concern is augmented when there is a history of a preceding infection.
- •
Patients with suspected HSP should have a 3- or 4-mm punch biopsy for routine hematoxylin and eosin (H&E) studies.
- •
Ideally, an additional punch biopsy, or a portion of a divided punch biopsy, should be submitted for direct immunofluorescent studies (see Fig. 20.1 ). This specimen can never be placed in formalin.
- •
A minimal laboratory evaluation should include a complete blood count (CBC) with differential, liver and renal function tests, urinalysis, stool guaiac testing, throat culture, and determination of the streptozyme level.
Treatment
- •
For patients with renal disease, a number of retrospective or uncontrolled studies have supported the use of pulse methylprednisolone, with or without cyclophosphamide, triple immunosuppressive therapy (prednisolone, cyclophosphamide, and dipyridamole), corticosteroids and azathioprine, plasma exchange, with or without immunosuppressive therapy, and intravenous (IV) immunoglobulin.
- •
In children with significant or deteriorating renal disease, treatment should consist of high-dose corticosteroid therapy and an immunosuppressive agent (e.g., azathioprine, cyclophosphamide, or mycophenolate mofetil). Consultation with a pediatric nephrologist is important.
Prognosis
In patients who develop renal disease, complete spontaneous remission occurs in about 50%. About 5% of patients with renal disease will develop a progressive course that can include renal failure.
Acute Hemorrhagic Edema of Infancy
ICD10 code D69.0
INTERNAL ETIOLOGY
Pathogenesis
Some consider acute hemorrhagic edema (AHE) a unique disease, whereas others consider it a variant of HSP. AHE is also referred to as Finkelstein or Seidlmayer disease. AHE is considered by some to be an entity separate from HSP because of its distinct clinical appearance, the typical lack of systemic symptoms, and the younger ages of affected patients. Like HSP, AHE is a small vessel vasculitis caused by circulating IgA-mediated immune complexes that are deposited in vessel walls. Precipitating factors include viral infections, bacterial infections (e.g., Streptococcus, Salmonella, and Shigella spp. and S. aureus ) and, rarely, medications.
Clinical Features
- •
Classically, AHE affects infants and young children, with most cases occurring in those younger than 2 years. It can rarely affect older children.
- •
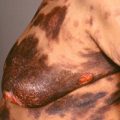
AHE consists of the abrupt onset of symmetrically distributed edematous plaques ( Figs. 20.5–20.7 ) that develop variable hemorrhage (see Figs. 20.6 and 20.7 ). Sometimes, the hemorrhage is subtle (see Fig. 20.5 ).
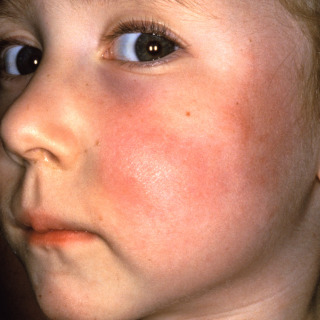
Fig. 20.5
Patient with acute hemorrhagic edema manifesting as an urticarial plaque with subtle hemorrhage.
(From the Fitzsimons Army Medical Center Collection, Aurora, CO.)
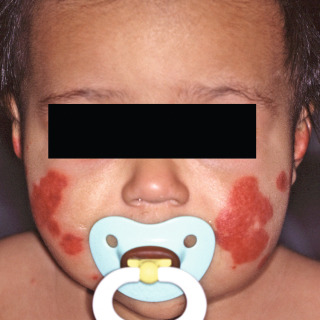
Fig. 20.6
Patient with developed case of acute hemorrhagic edema manifesting as an urticarial plaque with extensive hemorrhage.
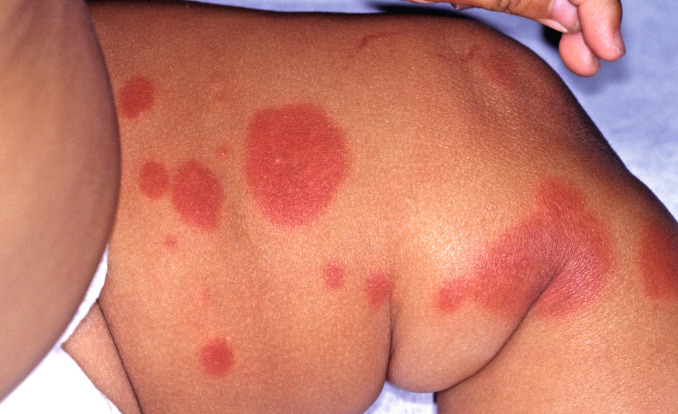
Fig. 20.7
Large urticarial plaques with extensive hemorrhage of the lower extremity in an infant.
(From the Joanna Burch Collection, Aurora, CO.)
- •
The face and lower extremities are usually affected, followed by the arms and buttocks. Truncal lesions are uncommon in AHE.
- •
Although arthritis-arthralgia, nephritis, and GI involvement have been reported, these conditions are uncommon in AHE but are common in HSP.
- •
Patients with AHE often have a low-grade fever.
Diagnosis
- •
An infant or young toddler with purpura in fixed urticarial lesions on the face and/or lower extremities should suggest AHE, especially if there is a history of a preceding infection. In Fig. 20.5 , the nasal secretions of an upper respiratory infection are readily evident.
- •
All patients with suspected AHE should have a 3- or 4-mm punch biopsy performed for routine H&E examination.
- •
Ideally, an additional punch biopsy, or a portion of a larger divided punch should be submitted for direct immunofluorescent studies. This specimen can never be placed in formalin.
- •
Most cases do not require further evaluation but, in select cases with signs of systemic disease, it may be wise to perform a CBC with differential, liver and renal function tests, urinalysis, stool guaiac testing, throat culture, and determination of streptozyme level.
Treatment
- •
In the absence of systemic findings, no treatment is necessary, because AHE is a self-limited disease without permanent sequelae.
- •
In patients with systemic findings, anecdotal cases have responded well to oral corticosteroids.
Prognosis
AHE typically resolves within 2 to 3 weeks, without treatment. There have been case reports of recurrent disease.
Leukocytoclastic (Hypersensitivity) Vasculitis
ICD10 code D69.0
INTERNAL ETIOLOGY
Pathogenesis
Although LCV is probably the more modern term, the older synonym, “hypersensitivity vasculitis” (HV) emphasizes that this small vessel vasculitis occurs as a reaction to a perturbation in homeostasis. LCV (HV) may occur for a variety of reasons, including infections (streptococcal infections), medication reactions (see box), certain ingestions, and collagen vascular disease. Urticarial vasculitis is another distinct subset of LCV (HV) (see Chapter 5 ). HV can be IgM-mediated (collagen vascular diseases), IgA-mediated (HSP) or, less often, IgG-mediated.
Clinical Features
- •
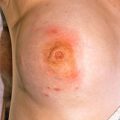
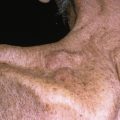
LCV (HV) presents with the abrupt onset of palpable purpura that mainly affects dependent areas of the lower legs, in particular.
- •
Most individual lesions of LCV (HV) are 1 to 6 mm in size, but larger lesions are not uncommon ( Figs. 20.8 and 20.9 ).
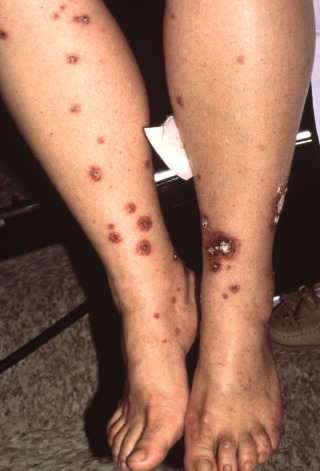
Fig. 20.8
Hydrochlorothiazide-induced adult hypersensitivity vasculitis. The patient had no evidence of systemic involvement.
(From the Fitzsimons Army Medical Center Collection, Aurora, CO.)
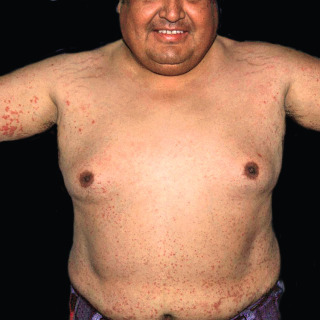
Fig. 20.9
Adult hypersensitivity vasculitis of unknown cause in an adult Hispanic man showing generalized involvement, including the upper arms and torso. The cause was never identified.
(From the Fitzsimons Army Medical Center Collection, Aurora, CO.)
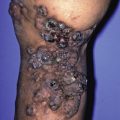
- •
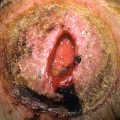
Clinical variants may include pustules, hemorrhagic vesicles ( Fig. 20.10 ), purpuric lesions with central necrosis, and lesions in a reticulated pattern.
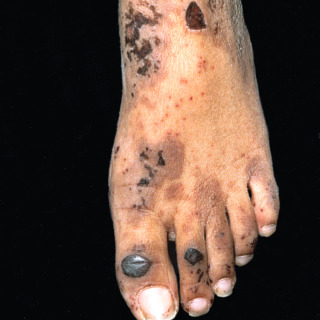
Fig. 20.10
Severe adult hypersensitivity vasculitis in an adult with large hemorrhagic bullous lesions and superficial ulcerations. The patient had arthralgias and mild renal involvement.
- •
Lesions may be asymptomatic, or a burning sensation or pain may be reported by some patients.
- •
Systemic symptoms may include fever, malaise, and arthralgias (the latter is most common).
- •
Most cases of LCV (HV) in adults are confined to the skin, but any organ system, including the kidneys, GI tract, joints (arthritis), lungs, and heart can be involved.
Diagnosis
- •
The sudden onset of palpable purpura on the lower extremities of an adult should raise suspicion for LCV (HV), and a history and review of systems should be carried out to identify a potential trigger.
- •
All cases of suspected LCV (HV) should have a 3- or 4-mm punch biopsy performed to include the subcutaneous fat. If possible, ulcerated or necrotic lesions should be avoided.
- •
An additional punch biopsy of perilesional skin should be submitted for direct immunofluorescent studies. This specimen cannot be placed in formalin.
- •
Reasonable additional laboratory screening studies may include a CBC, throat culture (for streptococcal infection), urinalysis (to exclude renal involvement), comprehensive metabolic panel (to include renal function studies and liver enzymes), and stool guaiac testing.
- •
Other studies, including testing for levels of cryoglobulin, complement (C1q, C3/C4, CH50), rheumatoid factor, antinuclear antibody (ANA), and extractable nuclear antigen antibodies (ENAs), HIV testing, hepatitis B and C studies, and chest x-ray may be ordered in certain cases.
Treatment
- •
Removal or treatment of the precipitating cause is always the initial step in management.
- •
Cases of small vessel vasculitis limited to the skin can be treated with prednisone (20–60 mg/day, with a taper over 2 to 3 weeks), colchicine (0.6 to 2.4 mg/day), and dapsone.
Clinical Course
Individual lesions of LCV (HV) heal over 2 to 4 weeks but may leave a reddish-brownish discoloration (hemosiderosis) or scar. Patients may have a single episode of LCV (HV) or multiple recurrent episodes. Rare patients may experience the continued development of new lesions without interruption.
Disseminated Gonococcal Infection
ICD10 code 154.9
BACTERIAL INFECTION
Introduction
In 2014, there were more than 350,000 cases of disseminated gonococcal infection (DGI) reported, with an estimated 300,000 additional cases unreported. DGI is due to intravascular septicemia caused by Neisseria gonorrhoeae . The bacterium gains access to the bloodstream from asymptomatic or untreated genital infections. DGI develops in 1% to 3% of all patients with gonorrhea, and the vast majority of cases occur in women. There is an increased risk of dissemination during menstruation, during pregnancy, or with pelvic operations or the placement of an intrauterine device. Patients with deficits in the later components of the complement pathway are particularly prone to DGI.
Clinical Presentation
- •
DGI usually occurs 1 week after menstruation or 2 to 3 weeks after the initial gonococcal infection.
- •
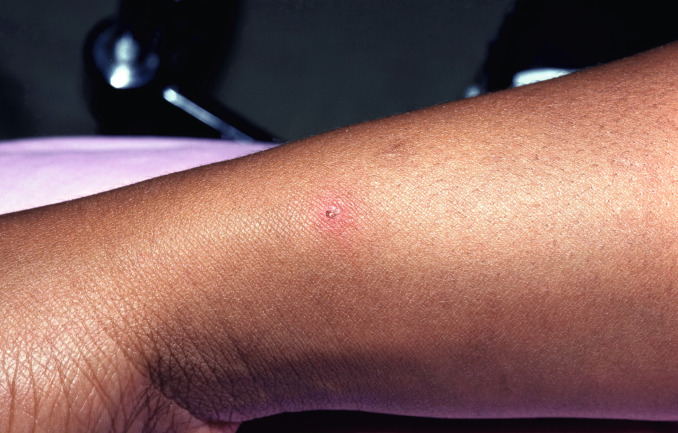
Cutaneous lesions are present in more than 60% of patients and consist of tender hemorrhagic lesions, usually on the arms and legs ( Figs. 20.11–20.13 ). A pustular component is not uncommon. Cutaneous lesions often present before arthritic symptoms.
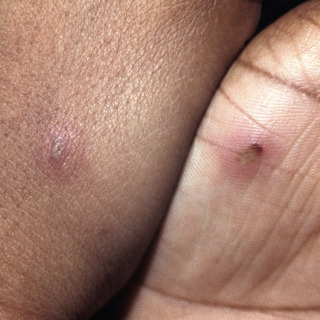
Fig. 20.11
Patient with disseminated gonococcal infection, with pustular and hemorrhagic lesions.
(From the Fitzsimons Army Medical Center Collection, Aurora, CO.)
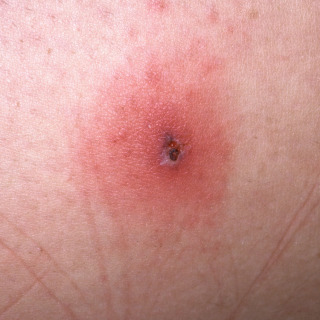
Fig. 20.12
Patient with disseminated gonococcal infection demonstrating papulonecrotic lesion with an intense rim of erythema.
(From the Fitzsimons Army Medical Center Collection, Aurora, CO.)