44 Reconstruction of urogenital defects
Congenital
Synopsis



Introduction
Bladder exstrophy tends to occur in nulliparous, young females’ offspring, with a recurrence risk of 1 : 275 and a risk of 1 : 70 in progeny. Patients have a low-set umbilicus and wide pubic diastasis. The pubic bone is shortened by 30%, with external rotation of the posterior pelvis. The clitoris or penis is short and bifid, and the urinary sphincter mechanism is underdeveloped. Nearly all patients have radiographic evidence of vesicoureteral reflux, and the incidence of inguinal hernias is increased. Treatment is discussed primarily in Volume 4, Chapter 13.
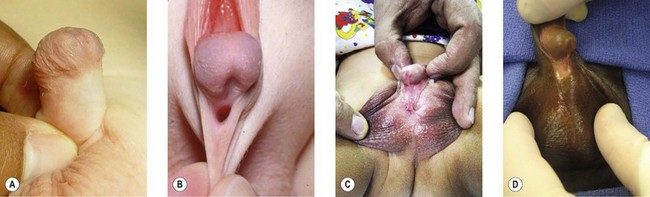
Hypospadias occurs when the development of the urethral spongiosum and prepuce halts, resulting in a meatal location proximal to that of the distal glans. A hypospadic meatus is often accompanied by penile chordee as the normal embryological correction of curvature is likewise arrested. The severity of hypospadias is dictated by meatal location, ranging from perineal to glanular (Figs 44.1, 44.2). The more severe the hypospadias, the more likely it is accompanied by chordee and penoscrotal transposition (Figs 44.3 and 44.4).
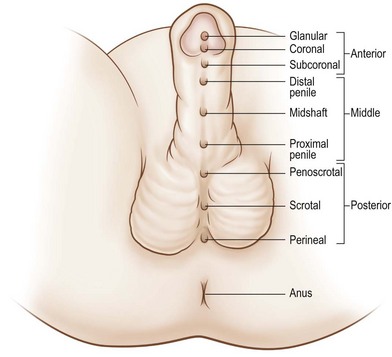
Fig. 44.1 Spectrum of hypospadias.
(Reproduced from Wein: Campbell-Walsh Urology, 9th ed. © 2007 Saunders.)

Historical perspective
Vaginal agenesis was first documented in the 16th century, with the first repairs described 300 years later. In 1898, Abbé1 described dissecting a neovaginal canal and lining it with split-thickness skin grafts situated over a temporary rubber stent. The procedure was popularized by McIndoe,2 who published a series of 63 such repairs. The usage of bowel, both small and large, as well as buccal mucosa and cultured vaginal tissue has been reported.3,4 Nonoperative approaches have also been employed, first through manual pressure and subsequently through the use of a modified seat-based dilation system.5,6 The resultant neovagina is lined by cutaneous squamous epithelium and requires external lubrication for intercourse.
The first description of bladder exstrophy repair was by Sweetser et al. in 1952.7 The first successful cloacal exstrophy repair was reported in 1960 by Rickham.8 The flourishing of neonatal intensive care over the last 50 years has resulted in near-universal survival for these infants.
Over 300 repairs for hypospadias have been described. Galen is credited with the first descriptions of hypospadias and chordee in the second century ad, with Vesalius, amongst others, elaborating on the disease description.9 The first millennium contained descriptions of amputation distal to the meatus with or without penile skin stretching; the second thousand years was characterized by tunneling and cannulation procedures in which a penile tunnel was repeatedly catheterized until it epithelialized.9 Many modern techniques have strong resemblance to procedures first described centuries ago. The concept of creating a tube in situ was documented in the 19th century by Anger,10 who overlapped tubularized flaps adjacent to the urethra, and Thiersch and Duplay,11 who incised tissue adjacent to the urethral groove, allowing for dorsal urethral closure. Duplay was also studying meatal advancement techniques. Pedicle skin grafts may be traced to Van Hook, who utilized a preputial pedicle flap in 1896.12 The utilization of free flaps was pioneered by Nové-Josserand,13 who wrapped a penile and preputial autograft around a catheter and tunneled the graft from glans to perineum. These concepts, all arising over 100 years ago, remain central to hypospadias repair today.
Basic science/disease process
Development
The cloaca appears during the second gestational week, with the urorectal septum forming during week 4 (Fig. 44.5). The urorectal septum fuses with the cloacal membrane by week 7. Defects of the cloacal membrane may result in bladder or cloacal exstrophy and epispadias. The mullerian ducts fuse to become the uterovaginal canal. Distally, the urogenital sinus forms the vestibule and the intervening sinovaginal bulbs canalize to form the distal vaginal canal. Paramesonephric abnormalities are frequently associated with ipsilateral renal anomalies, partly based upon their proximity during development. Anomalies of agenesis and fusion may occur. When the urogenital sinus fails to develop into the distal vagina, atresia results; agenesis occurs when the proximal third of the vagina fails to develop in a 46XX phenotypic female. Transverse septa arise from failure of fusion or canalization of the urogenital sinus and mullerian ducts. Meyer–Rokitansky–Kusler–Hauser syndrome consists of mullerian aplasia, often accompanied by renal and cervicothoracic dysplasias.
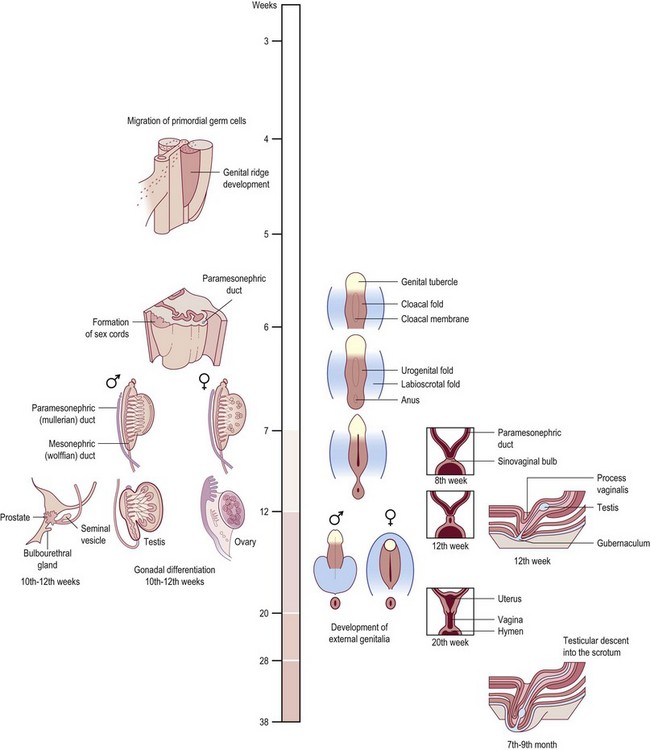
Fig. 44.5 Development of the urogenital tract.
(Modified from Larsen WJ: Human Embryology. New York, Churchill Livingstone, 1997.)
Between weeks 7 and 8 of gestation, the male gonads differentiate secondary to the transcription of the SRY gene, thereby producing testosterone. The distance between anus and genitalia increases, the phallus elongates, the genital folds fuse, the preputial folds form and fuse dorsally, and during the 11th gestational week, the urethral folds fuse ventrally. If the genital folds fail to fuse, as in hypospadias, the preputial folds also fail to fuse ventrally, resulting in excessive dorsal preputial tissue. By week 16, the glandular urethra forms. The glandular portion of the urethra is lined by squamous epithelium which likely also arises from urogenital sinus origin, but then undergoes differentiation.14 Histologically, the hypospadic urethral plate contains sinusoids of urethral spongiosum and no scar tissue. The innervation of the hypospadic penis is like that of the normal penis: the pedendal nerve gives off the dorsal nerve which travels as two bundles on either side of 12 o’clock.
Diagnosis/patient presentation
Patient selection
Patients presenting with the exstrophy–epispadias complex are managed with a staged procedure.15 Bladder and abdominal closures with bilateral osteotomy are performed in the newborn period. Epispadias repair occurs 6–12 months later, and bladder neck reconstruction with ureteral reimplantation in another 4–5 years if indicated.
The optimal timing for hypospadias repair is 6 months of age in a full-term male. Ninety-two percent of pediatric urologists perform a TIP procedure for distal hypospadias .13 Proximal hypospadias without curvature is repaired by TIP or onlay island flap by equal proportions of urologists, while proximal hypospadias with severe chordee is most often repaired by a staged repair.16 Intramuscular testosterone injections of 25 mg/dose or 2 mg/kg/dose may be administered up to three times prior to repair in an attempt to improve penile vascularity and tissue robustness, thereby facilitating subsequent surgery.
Treatment/surgical technique
Vaginal reconstruction
Vaginal agenesis
The use of bowel results in mucus production which may require daily douching and result in odor or ulcerations.17 While bowel preparation has historically been advised, the authors have found it to be unnecessary.18,19 A Pfannenstiel incision is appropriate, although a midline incision may also be employed. A 10-cm segment of mobile sigmoid or ileum may be taken out of continuity and anastomosed directly to the skin dimple (Fig. 44.6). While some surgeons detubularize and subsequently fold the bowel to form the neovagina, the authors find this unnecessary and perform a simple proximal-limb closure with 2-0 absorbable suture. The distal aspect of the bowel limb is anastomosed in recessed fashion to skin flaps or the rudimentary vagina is cases of proximal atresia. The bowel anastomosis may be hand-sewn or stapled.
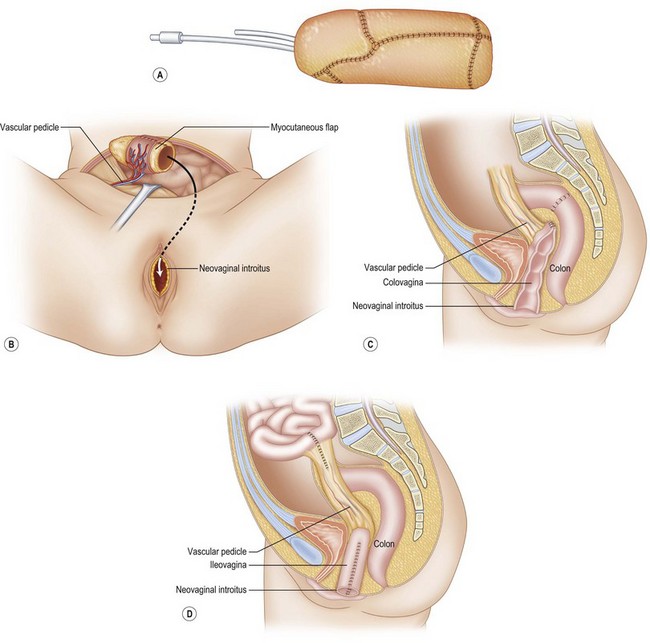
Within bowel constructs, stenosis occurs more often with ileum than with colon.20

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


