Plectin is an important organizer of the keratin filament cytoskeleton in basal keratinocytes. It is essential for anchoring these filaments to the extracellular matrix via hemidesmosomal integrins. Loss of plectin or incorrect function of the protein due to mutations in its gene can lead to various forms of the skin blistering disease, epidermolysis bullosa simplex. Severity and subtype of the disease is dependent on the specific mutation and can be associated with (late-onset) muscular dystrophy or pyloric atresia. Mouse models mimicking the human phenotypes allow detailed study of plectin function.
Within cells, multiple cytoskeletal networks (built from actin, intermediate filament (IF) subunit proteins, or tubulin) cooperate with each other to perform their diverse tasks. Accessory proteins are essential for the controlled spatiotemporal assembly and interaction of cytoskeletal filaments, their connection with various cellular components, and the generation of movement. An important family of structurally and, in part, functionally related proteins capable of interlinking different elements of the cytoskeleton are the plakins or cytolinkers. Plakins are large proteins that were first identified on the basis of their association with IFs or IF-anchoring structures, such as desmosomes and hemidesmosomes (HDs). Thus, cytolinkers play crucial roles in maintaining cell and tissue integrity and orchestrating dynamic changes in cytoarchitecture and cell shape. With their multidomain structure and enormous size, they serve as scaffolding platforms for the assembly, positioning, and regulation of signaling complexes. Among the plakins, plectin is the best characterized and seems the most versatile.
Gene and protein structure
Full-length plectin has a deduced molecular mass ranging from 499 to 533 kDa, depending on the particular isoform. Rotary shadowing electron microscopy of purified plectin molecules revealed a dumbbell-like structure comprising a central 200-nm–long rod domain flanked by large globular domains ( Fig. 1 A). The multidomain structure of plectin was confirmed by secondary structure predictions based on complementary DNA and deduced amino acid sequences. These analyses also revealed that the rod domain is characterized by long stretches of heptad repeats, distinctive of α-helical coiled coils, and exhibits a staggered strict period of 10.4 for acidic and basic residues, suggesting an energetically most favored parallel arrangement of a plectin dimer. This is supported by the microscopic dimensions of plectin and gel permeation high-performance liquid chromatography data indicating a molecular mass of plectin molecules in solution of slightly more than 1.1 × 10 6 Da. The structure of the 214-kDa carboxy (C)-terminal globular domain, encoded by a single very large (>6 kb) exon, is dominated by six highly homologous, approximately 300 amino acid residues–long repeat domains, which also occur in different numbers in other cytolinker (plakin) protein family members, such as desmoplakin, the epithelial and neuronal isoforms of BPAG1/dystonin, envoplakin, and epiplakin. The amino (N)-terminal domain comprises an actin-binding domain (ABD), shared by a large superfamily of actin-binding proteins, and a plakin domain containing several spectrin repeats interrupted by an SH3 domain.
Plectin is expressed as several protein isoforms from a single gene, PLEC1, that is located on chromosome 8q24 in humans (see Fig. 1 C). The plectin gene locus in mouse (chromosome 15) has been analyzed in detail, revealing a genomic exon-intron organization with more than 40 exons spanning 62 kb on chromosome 15 and an unusual 5′ transcript complexity of plectin isoforms. Eleven exons (1–1j) have been identified that alternatively splice directly into a common exon 2 (which is the first exon to encode plectin’s highly conserved ABD), three (−1, 0a, and 0) that are spliced upstream of exon 1c, and two additional ones (2α and 3α) that are optionally spliced within the ABD-encoding exons (2–8; see Fig. 1 D). In a recent study, the human and rat gene loci were reanalyzed based on newly available genome sequences and compared to the mouse gene. In rat, all 11 alternative first exons identified in mouse were found, whereas in the human gene, so far, the presence of eight of these exons has been confirmed. Furthermore, a rodless human plectin variant, as judged by molecular size, has been detected on the protein level.
Expression, subcellular localization, and binding partners
Plectin is expressed in virtually all mammalian tissues and cell types, as has been demonstrated using antisera and a panel of monoclonal antibodies. It is particularly prominent in various types of muscle, stratified and simple epithelia, cells forming the blood-brain barrier, and tissue layers at the interface between tissues and fluid-filled cavities, where it was found at the surfaces of kidney glomeruli, liver bile canaliculi, bladder urothelium, gut villi, ependymal layers lining the cavities of brain and spinal cord, and endothelial cells of blood vessels. At the cellular level, plectin codistributes with different types of IFs and is located at plasma membrane attachment sites of IFs and microfilaments, such as HDs, desmosomes, Z-line structures and dense plaques of striated and smooth muscle, intercalated discs of cardiac muscle, and focal contacts.
Ribonuclease protection experiments performed on a panel of mouse tissues and cell types, using antisense riboprobes specific for the various alternative first exons of plectin, provided clear evidence for the occurrence of tissue-specific or dominant plectin isoforms. For example, plectin transcripts containing exon 1d were exclusively found in skeletal and heart muscle, whereas exon 1a–containing transcripts were dominant in organs rich in epithelial cell types, such as lung, small intestine, and, in particular, skin. Tissue-specific expression was characteristic also of the two optionally spliced exons, 2α and 3α, with isoforms containing exon 2α being expressed in brain, heart, and skeletal muscle, and exon 3α being brain-specific. Furthermore, the alternative first exons were found to determine the stability of gene products by controlling initiation of translation. The most intriguing finding, however, was that the short alternative N-terminal sequences encoded by the different first exons direct the various isoforms to distinct subcellular locations. For instance, plectin 1a specifically associated with HD-like structures in keratinocytes, plectin 1b was found associated exclusively with mitochondria, and plectin 1f was concentrated in vinculin-positive structures at actin stress fiber ends. Thus, it seems that each cell type (tissue) contains a unique set (proportion and composition) of plectin isoforms, as if custom-made for specific requirements of the particular cells. Concordantly, individual isoforms were found to carry out distinct and specific functions.
Consistent with its varied subcellular localization, plectin has been shown to directly interact with a variety of cytoskeletal structures and proteins. In its C-terminal domain, plectin contains a multifunctional IF-binding site that was mapped to an approximately 50 amino acid–long sequence located between the highly conserved core regions of its C-terminal repeats 5 and 6. This site mediates binding to several types of IF subunit proteins, including vimentin, desmin, glial fibrillary acidic protein (GFAP), the nuclear IF protein lamin B, and type I and type II cytokeratins. A highly conserved ABD close to its N terminus mediates binding not only to actin but also to the integrin subunit β4, vimentin, the EF-ZZ domain of dystrophin and utrophin, and binding sites for nesprin-3. Additionally, the signaling molecule phosphatidylinositol-4,5-bisphosphate (PIP 2 ) binds within the ABD, regulating binding of plectin to actin. Binding of integrin β4 and actin to plectin’s ABD has been shown to be mutually exclusive. Furthermore, the interactions of actin and integrin β4 with plectin isoform 1a are regulated in a Ca 2+ -dependent manner by calmodulin, which binds to the CH1 domain of the ABD. Additional binding sites for integrin β4 and for β-dystroglycan are contained in the plakin and C-terminal domains of plectin. Direct interactions of plectin with the membrane skeleton proteins fodrin and α-spectrin, the desmosomal protein desmoplakin, and microtubule-associated proteins (MAP2 and MAP1 subtypes) from brain have been reported. Furthermore, whole-mount electron microscopy was used to demonstrate that plectin is capable of physically linking IFs to microtubules.
Recently, it has been shown that plectin not only functions as a cytolinker but also acts as a scaffold for proteins involved in signaling, anchoring them at specific sites within a cell. Such proteins include the nonreceptor tyrosine kinase Fer ; the receptor for activated C kinase 1 (RACK1) that is sequestered to the cytoskeleton through plectin during initial stages of cell adhesion ; and the γ1 subunit of the energy-controlling AMP-activated protein kinase.
Expression, subcellular localization, and binding partners
Plectin is expressed in virtually all mammalian tissues and cell types, as has been demonstrated using antisera and a panel of monoclonal antibodies. It is particularly prominent in various types of muscle, stratified and simple epithelia, cells forming the blood-brain barrier, and tissue layers at the interface between tissues and fluid-filled cavities, where it was found at the surfaces of kidney glomeruli, liver bile canaliculi, bladder urothelium, gut villi, ependymal layers lining the cavities of brain and spinal cord, and endothelial cells of blood vessels. At the cellular level, plectin codistributes with different types of IFs and is located at plasma membrane attachment sites of IFs and microfilaments, such as HDs, desmosomes, Z-line structures and dense plaques of striated and smooth muscle, intercalated discs of cardiac muscle, and focal contacts.
Ribonuclease protection experiments performed on a panel of mouse tissues and cell types, using antisense riboprobes specific for the various alternative first exons of plectin, provided clear evidence for the occurrence of tissue-specific or dominant plectin isoforms. For example, plectin transcripts containing exon 1d were exclusively found in skeletal and heart muscle, whereas exon 1a–containing transcripts were dominant in organs rich in epithelial cell types, such as lung, small intestine, and, in particular, skin. Tissue-specific expression was characteristic also of the two optionally spliced exons, 2α and 3α, with isoforms containing exon 2α being expressed in brain, heart, and skeletal muscle, and exon 3α being brain-specific. Furthermore, the alternative first exons were found to determine the stability of gene products by controlling initiation of translation. The most intriguing finding, however, was that the short alternative N-terminal sequences encoded by the different first exons direct the various isoforms to distinct subcellular locations. For instance, plectin 1a specifically associated with HD-like structures in keratinocytes, plectin 1b was found associated exclusively with mitochondria, and plectin 1f was concentrated in vinculin-positive structures at actin stress fiber ends. Thus, it seems that each cell type (tissue) contains a unique set (proportion and composition) of plectin isoforms, as if custom-made for specific requirements of the particular cells. Concordantly, individual isoforms were found to carry out distinct and specific functions.
Consistent with its varied subcellular localization, plectin has been shown to directly interact with a variety of cytoskeletal structures and proteins. In its C-terminal domain, plectin contains a multifunctional IF-binding site that was mapped to an approximately 50 amino acid–long sequence located between the highly conserved core regions of its C-terminal repeats 5 and 6. This site mediates binding to several types of IF subunit proteins, including vimentin, desmin, glial fibrillary acidic protein (GFAP), the nuclear IF protein lamin B, and type I and type II cytokeratins. A highly conserved ABD close to its N terminus mediates binding not only to actin but also to the integrin subunit β4, vimentin, the EF-ZZ domain of dystrophin and utrophin, and binding sites for nesprin-3. Additionally, the signaling molecule phosphatidylinositol-4,5-bisphosphate (PIP 2 ) binds within the ABD, regulating binding of plectin to actin. Binding of integrin β4 and actin to plectin’s ABD has been shown to be mutually exclusive. Furthermore, the interactions of actin and integrin β4 with plectin isoform 1a are regulated in a Ca 2+ -dependent manner by calmodulin, which binds to the CH1 domain of the ABD. Additional binding sites for integrin β4 and for β-dystroglycan are contained in the plakin and C-terminal domains of plectin. Direct interactions of plectin with the membrane skeleton proteins fodrin and α-spectrin, the desmosomal protein desmoplakin, and microtubule-associated proteins (MAP2 and MAP1 subtypes) from brain have been reported. Furthermore, whole-mount electron microscopy was used to demonstrate that plectin is capable of physically linking IFs to microtubules.
Recently, it has been shown that plectin not only functions as a cytolinker but also acts as a scaffold for proteins involved in signaling, anchoring them at specific sites within a cell. Such proteins include the nonreceptor tyrosine kinase Fer ; the receptor for activated C kinase 1 (RACK1) that is sequestered to the cytoskeleton through plectin during initial stages of cell adhesion ; and the γ1 subunit of the energy-controlling AMP-activated protein kinase.
Plectin gene mutations and epidermolysis bullosa simplex
In 1996, several groups reported that patients suffering from epidermolysis bullosa simplex with muscular dystrophy (EBS-MD), an autosomal recessive disorder with neonatal skin blistering and delayed, progressive muscular weakness, lacked plectin expression in skin and muscle tissues due to defects in the plectin gene. Based on plectin’s prominence at plasma membrane junctional sites of IFs, it had been anticipated that defects in its gene would lead to such phenotypes. The direct interaction of plectin with the HD integrin subunit β4, generating a linkage between the IF cytoskeleton and the extracellular matrix (see Fig. 1 F), provided a molecular model for the skin blistering phenotype. In many cases, the responsible mutations were nonsense mutations, out-of-frame insertions, or deletions within exons 31 or 32, resulting in premature stop codons. This predicts truncated polypeptides and mRNA down-regulation through nonsense-mediated mRNA degradation. Lack of immunoreactivity of samples from such patients suggested the absence of plectin molecules. As the antibodies most commonly used were immunoreactive with the rod or C-terminal domains, however, truncated versions of plectin might still have existed. One notable mutation is 13480ins16 (homozygous), where normal levels of mRNA were found and protein was detectable, although at reduced levels. EBS-MD was further found to be caused by several in-frame insertion and deletion mutations, such as homozygous 2674del9 [exon 21] or compound heterozygous 1537ins36/2674del9 [exons 14 and 21]. In general, the phenotypes of EBS-MD patients vary considerably in severity of skin blistering and onset of muscular dystrophy.
EBS with pyloric atresia (EBS-PA) is another HD variant that manifests as severe neonatal skin blistering and gastric abnormalities (pyloric or duodenal atresia) and frequently leads to early postnatal demise of affected individuals. Plectin mutations underlying EBS-PA comprise homozygous or compound heterozygous nonsense mutations and, in one case, a homozygous 21-bp in-frame deletion within exon 22. Attempts to correlate plectin genotypes with the EBS-PA versus EBS-MD phenotypes could not provide any clues for the intriguing phenotypic differences. So far, however, no mutations causing PA have been found within the rod-encoding exon 31. Thus, the presence of rodless splice variants of plectin, which have been detected in some EBS-MD patients, may be the reason for the less severe phenotype compared to EBS-PA.
Besides the recessive forms of plectin-linked EBS, one autosomal dominant form of EBS, designated EBS-Ogna, has been identified. This rare mutation was identified in a kindred near the small Norwegian town of Ogna (hence its name) and independently in a German family. It constitutes a heterozygous missense mutation, R2000W, within the rod-encoding exon 31, apparently perturbing the function of plectin through dominant negative interference. These patients do not develop MD or show any signs of PA.
The position numbering of mutations found in the existing literature is based on different sequence database entries, and thus is confusing. The reason for this is the alternative 5′ splicing of plectin isoforms, each with its own database entry. Therefore, the authors have compiled a list of all plectin mutations published thus far ( Table 1 ) and assigned numbers to the mutations based on a common reference sequence (accession no. NM_000445), plectin isoform 1c (also described as variant 1 in the databases). The positions of the mutations and resulting phenotypes are depicted in Fig. 1 B. Thus far, no mutations in the alternative first exons have been described. Mutations that would lead to the elimination of only one specific isoform, however, are expected to be found in the future. Likely candidates are exon 1a (skin phenotype) and 1d (muscle phenotype). The authors propose that such mutations are named based on the corresponding database sequence (see Fig. 1 E) and that the isoform or variant be indicated in that case (eg, a hypothetical mutation in exon 1a would be named R7X 1a ).
| Mutation a | Original Designation b | Exon c | Genotype d | Case e | References |
|---|---|---|---|---|---|
| EBS-MD | |||||
| 963ins3 | 1287ins3, 1008ins3 | 9 | c.het. (Q1408X) | 14 | |
| 1537ins36 | 1541ins36 | 14 | c.het. (2674del9) | 20 | |
| 2674del9 | 2677del9, 2719del9 | 21 | hom. | 4,20 | |
| 2700-9del21 | 2745-9del21 | 22 | c.het. (5038delG) | 12 | |
| Q1053X | (Same) | 24 | c.het. (Q1936X) | 9 | |
| Q1408X | (Same), Q1518X | 31 | c.het. (963ins3) | 14 | |
| 4313ins13 | 4359ins13 | 31 | c.het. (4371delC) | 8 | |
| 4371delC | 4416delC | 31 | c.het. (4313ins13) | 8 | |
| 5024del19 | 5069del19 | 31 | hom. | 6 | |
| 5038delG | 5083delG | 31 | c.het. (2700-9del21) | 12 | |
| 5103del8 | 5148del8 | 31 | hom. | 1 | |
| Q1713X | (Same) | 31 | c.het. (R2351X) | 11 | |
| 5264insG | 5309insG, 5588insG | 31 | hom. | 16,19 | |
| Q1910X | (Same) | 31 | hom. | 5 | |
| Q1936X | (Same) | 31 | c.het. (Q1053X) | 9 | |
| 5821delC | 5866delC | 31 | hom. | 3 | |
| 5854ins8 | 5907ins8 | 31 | hom. | 2 | |
| 5860del2 | 5905del2 | 31 | hom. | 7 | |
| E2005X | (Same) | 31 | c.het. (K4460X) | 13 | |
| R2319X | (Same) | 31 | hom. | 18 | |
| R2351X | (Same) | 31 | c.het. (Q1665) | 11 | |
| R2421X | (Same) | 31 | c.het. (12588ins4) | 10,22 | |
| R2465X | (Same) | 31 | hom. | 17 | |
| 12588ins4 | 12633ins4 | 32 | c.het. (R2421X) | 10 | |
| K4460X | (Same) | 32 | c.het. (E2005X) | 13 | |
| 13480ins16 | 13803ins16 | 32 | hom. | 15 | |
| EBS-PA | |||||
| Q305X | (Same) | 9 | hom.; c.het. (1350G>A) | 23,28 | |
| 1350G>A | 1344G>A | 12 | c.het. (Q305X) | 23 | |
| 1569del4 | 1563del4, 1567del4 | 14 | hom. | 27 | |
| 2686del14 | 2727del14 | 21 | hom. | 22 | |
| 2775del21 | 2769del21 | 22 | hom. | 26 | |
| R1189X | (Same) | 27 | hom.; c.het. (Q2538X) | 24 | |
| Q2538X | (Same) | 32 | c.het. (R1189X) | 24 | |
| R3029X | (Same) | 32 | hom. | 25 | |
| EBS-Ogna | |||||
| R2000W | R2110W | 31 | het. (dominant) | 29,30 | |
Related posts:
 Mutation Mechanisms
Mutation Mechanisms
 Adhesion and Migration, the Diverse Functions of the Laminin α3 Subunit
Adhesion and Migration, the Diverse Functions of the Laminin α3 Subunit
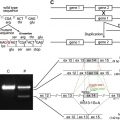
 Laryngo-onycho-cutaneous Syndrome
Laryngo-onycho-cutaneous Syndrome
 Ectodermal Dysplasia-Skin Fragility Syndrome
Ectodermal Dysplasia-Skin Fragility Syndrome
 Lethal Acantholytic Epidermolysis Bullosa
Lethal Acantholytic Epidermolysis Bullosa
 Understanding the Pathogenesis of Recessive Dystrophic Epidermolysis Bullosa Squamous Cell Carcinoma
Understanding the Pathogenesis of Recessive Dystrophic Epidermolysis Bullosa Squamous Cell Carcinoma
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


