Epidermolysis bullosa acquisita (EBA) is an autoimmune blistering skin disease characterized by autoantibodies to type VII collagen. Clinically, a noninflammatory and an inflammatory variant of EBA can be distinguished. Despite major achievements in the understanding of EBA, current therapeutic options are far from optimal. However, with an emerging and more detailed understanding of the events ultimately leading to blister formation in EBA, novel therapeutic options may become available for patients with EBA. Therefore, this article reviews the current understanding of the pathogenesis of EBA and may indicate possible avenues towards a more targeted therapy for EBA and possibly other antibody-mediated autoimmune diseases.
In 1904, the term epidermolysis bullosa acquisita (EBA) was proposed as a descriptive clinical diagnosis for patients with adult onset and features resembling those of hereditary dystrophic epidermolysis bullosa. Almost 70 years later, EBA was distinguished from other bullous diseases based on distinctive clinical and histologic features, implementing the first diagnostic criteria for the disease. Since then, the understanding of the clinical presentation, histopathologic features, and pathogenesis of EBA has significantly increased. Despite these insights, therapy for patients with EBA still relies on general immunosuppression and overall remains unsatisfactory.
Loss of tolerance to type VII collagen
Identification of Type VII Collagen as the Autoantigen in EBA
In 1984, a skin basement membrane component was identified as the target of the autoantibody response in EBA. Subsequently, autoantibody specificity of sera from patients with EBA was mapped to the noncollagenous (NC) 1 domain of type VII collagen (COL7). Fine epitope mapping studies indicated that sera from patients with EBA bind to numerous antigens located within the NC1 domain ( Fig. 1 ). More specifically, major antigenic sites recognized by antibodies from patients with EBA clustered on fibronectin type (Fn) III–like domains and the von Willebrand factor domain. After successful expression of the cartilage matrix protein (CMP) domain of COL7, this domain was identified to also harbor major antigenic sites. Recent reports have indicated that some patients with EBA also recognize epitopes located within the NC2 domain or the triple-helical collagenous domain of COL7 (see Fig. 1 ). Further fine mapping studies identified octapeptide sequences within the NC1 domain as the major binding sites of anti-COL7 antibodies. However, in contrast to previous work, autoantibody binding was shown to be restricted to certain epitopes within the NC1 domain, which may be due to a relatively small sample size.
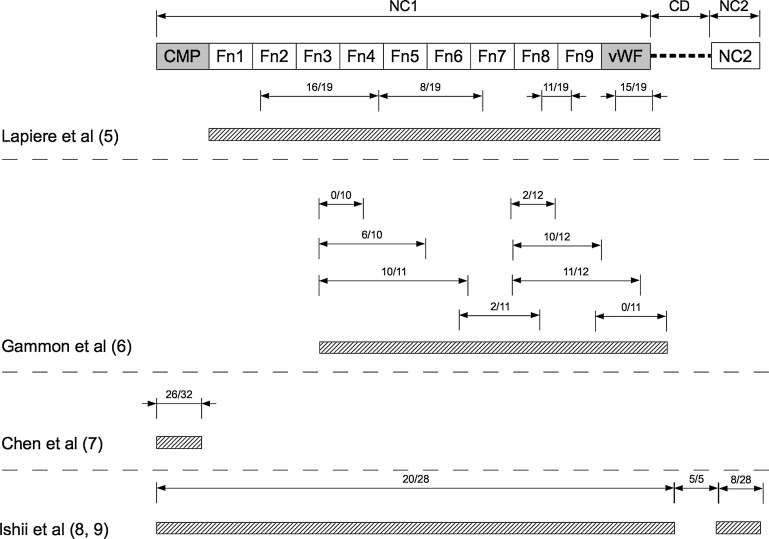
The pathogenic relevance of anti-COL7 antibodies has been demonstrated both in vitro and in vivo: in the presence of neutrophils, serum from patients with EBA induces dermal-epidermal separation when incubated with cryosections of human skin. This blister-inducing potential is retained if immunoglobulin G (IgG) from the serum is affinity purified using recombinant NC1. In contrast, the corresponding flow-through fraction fails to induce dermal-epidermal separation. When injected into mice, either human or rabbit anti-COL7 IgG elicits skin lesions, resembling those observed in human patients with EBA. In these anti-COL7 IgG transfer models of EBA, autoantibodies were directed against different epitopes within the murine NC1 domain, for example, the CMP domain or various Fn III–like domains. In addition, the disease can be induced in mice by immunization with an immunodominant peptide within the murine NC1 domain, which includes Fn III–like domains 7 to 9. These regions correspond to the similar Fn III–like domains of human COL7 (see Fig. 1 ).
Susceptibility to EBA is Linked to Major Histocompatibility Complex Genes
The major histocompatibility complex (MHC) or human leukocyte antigen (HLA) region is one of the most extensively studied regions in the genome because of the contribution of multiple variants of this locus to inflammatory diseases, including autoimmune disorders. In contrast to most autoimmune diseases, little is known about the susceptibility genes in EBA. In a small cohort of human patients, an association with HLA-DR2 has been described. Experimental data on immunization-induced EBA in mice support the notion that EBA is associated with MHC genes. In the initial description of this model, immunization with a recombinant fragment of murine COL7 led to clinical disease in 82% of SJL/J mice (H2s), whereas incidence in Balb/c mice (H2d) was significantly lower and outbreed SKH-1 mice were completely protected from EBA induction. Following up on this initial observation, detailed analyses including different inbreed mouse strains showed a clear association of EBA susceptibility with the H2s haplotype. Whereas 75% of mice carrying H2s (SJL/J, C57Bl/10.s) developed clinical EBA lesions, only 5% of inbreed non-H2s mouse strains were prone to EBA. The observation of clinical EBA in C57Bl/10.s mice, but not in genetically identical (with the exception of the MHC locus) C57Bl/10.q mice, underscores the importance of MHC in controlling tolerance to COL7. As murine and human MHC regions share little similarity, direct comparisons of genetic regions within this locus controlling EBA susceptibility cannot be made. However, both observations underscore the importance of the MHC locus in controlling autoimmune diseases such as EBA.
In addition to MHC genes, these experimental studies also indicated that genes outside the MHC locus have a strong influence in the control of tolerance to COL7. More specifically, the otherwise EBA-resistant C57Bl/6 mice become susceptible to EBA development when lacking expression of the inhibitory FcγRIIB receptor. In contrast to SJL/J mice, EBA in C57Bl/10.s mice is mild and mostly transient. Unpublished data from our laboratory, using a family of mice from a large, autoimmune-prone intercross line, has identified non-MHC genes, controlling susceptibility to immunization-induced EBA (unpublished data, 2011).
T Cells are Required for Induction of Experimental EBA
Induction of autoimmune diseases in experimental animals is generally a T-cell–dependent process. However, recent studies in the models of lupus erythematosus have indicated that under certain experimental settings, B cells, independent of T cells, are sufficient for the development of systemic autoimmunity. In EBA, T-cell reactivity to COL7 has been reported in individual patients. For example, T-cell reactivity against COL7 paralleled the clinical activity, preceding a delayed response of COL7-specific IgG. Furthermore, by enzyme-linked immunosorbent spot analysis, interleukin (IL)-5–secreting T h 2 cells, interferon-γ–secreting T h 1 cells, and an IL-10–secreting T-cell subset were detected on ex vivo stimulation of peripheral blood mononuclear cells from patients with EBA. However, the few numbers of patients with EBA investigated does not yet allow further insights into T-cell activation in patients with EBA. The critical contribution of T cells to the pathogenesis of experimental EBA has been demonstrated by recent data obtained from mice immunization-induced EBA, where genetic lack of T cells protected from disease induction. Furthermore, reconstitution of SJL nude mice with lymphocytes from wild-type SJL/J mice restored the susceptibility to immunization-induced EBA.
Conclusions and Remaining Questions Regarding the Loss of Tolerance
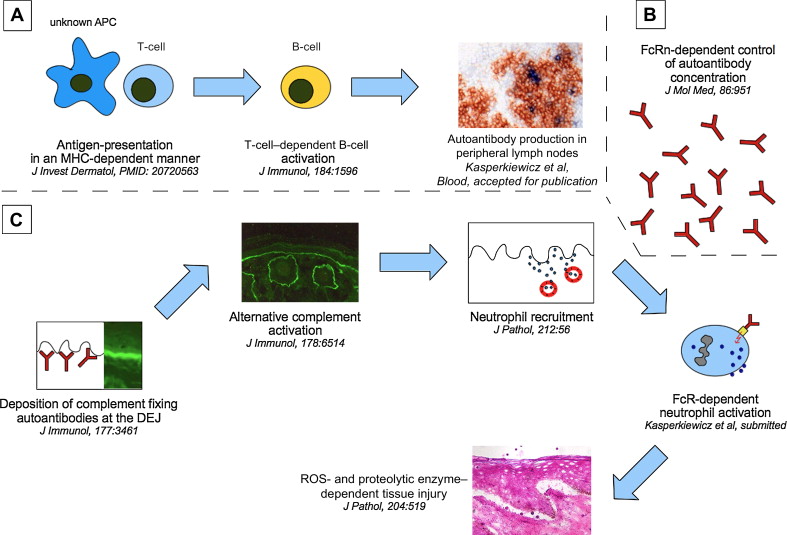
The pathogenic relevance of COL7 as the autoantigen in EBA has been convincingly demonstrated. Data from observational studies in humans with EBA and in model systems of EBA have indicated that the loss of tolerance to COL7 requires T cells and that this process is associated with specific genes ( Fig. 2 ). However, several questions need to be addressed for a detailed understanding of the pathogenesis of EBA:
- 1.
Is a distinct autoantibody specificity associated with the different EBA phenotypes?
- 2.
Which cells (if any), in addition to T cells, are involved in the loss of tolerance to COL7?
- 3.
Which individual genes are associated with EBA?
Autoantibody-induced Tissue Injury in EBA
After mounting an autoantibody response toward COL7, autoantibodies rapidly bind to their targeted antigen in the skin. For example, anti-COL7 IgG deposition is observed at the dermal-epidermal junction within 2 hours after subcutaneous injection at distant sites. Intravenously injected anti-COL7 IgG is detected within minutes at the dermal-epidermal junction (Ludwig and Zillikens, unpublished, 2011). After deposition at the dermal-epidermal junction, anti-COL7 autoantibodies can be detected by direct immunofluorescent microscopy for as long as 8 weeks. Anti-COL7 autoantibodies cross the placenta and can be detected in the offspring of the mice with experimental EBA and the neonates of female patients with EBA.
Autoantibody-induced Tissue Injury Depends on Fc-dependent Mechanisms
Without exception, autoantibody-induced tissue injury in all available experimental models of EBA depends on the Fc fragment of IgG. Incubation of human skin with anti-NC1 polyclonal IgG isolated from patients with EBA or a monoclonal antibody directed to an epitope on the central portion of the NC1 domain (clone LH7.2) leads to dermal-epidermal separation in the presence of neutrophils. Removal of the Fc portion of the autoantibodies by pepsin digestion abolished this pathogenic activity. Correspondingly, mice injected with either human or rabbit anti-COL7 IgG developed clinical lesions when injected with IgG but not when F(ab) 2 fragments were used at equimolar doses. In addition, chicken immunoglobulin Y (IgY), which is unable to activate murine complement and does not bind to murine Fc receptors, induced neither dermal-epidermal separation ex vivo, nor clinical EBA lesions in mice.
Distinct Autoantibody Isotypes Mediate Tissue Injury in EBA
However, effector functions of the Fc portion differ among isotypes and subclasses of antibodies. Depending on the subclass, antibodies greatly vary regarding their affinity to inhibiting and activating Fc receptors. In patients with EBA, anti-COL7 antibodies of all IgG subclasses can be detected with the following detection frequency: IgG4>IgG1>IgG2>IgG3. IgG subclasses of circulating anti-COL7 autoantibodies have not been demonstrated to correlate with the different clinical variants of EBA. However, recent work has demonstrated that only IgG1- and IgG3-anti-COL7 autoantibodies are capable to fix complement and induce dermal-epidermal separation ex vivo. This experimental model requires neutrophils for induction of dermal-epidermal separation, thus reflecting the inflammatory variant of EBA. The role of IgG2- and IgG4-anti-COL7 antibodies in the pathogenesis of EBA (especially the noninflammatory manifestation) cannot be excluded.
Activation of the Alternative Complement Cascade is Required in the Experimental Models of EBA
Injection of rabbit anti-mouse COL7 IgG into mice (passive EBA mouse model) elicits clinical EBA lesions, mimicking the inflammatory variant of the disease. In this model, mice lacking C5 are completely protected from disease induction. In line, chicken IgY did not induce clinical EBA lesions in mice. Further experiments demonstrated a predominant role of the alternative complement pathway, leading to the conversion of C5 to C5a, in lesion formation in this mouse model of EBA. In detail, factor B–deficient mice developed a delayed and significantly less severe blistering disease compared with control mice. Furthermore, local reconstitution of factor B–deficient mice with wild-type neutrophils reestablished disease susceptibility. Mice with defects in the classical- or lectin-complement activation pathways (C1q- or mannose binding lectin–deficient mice, respectively) showed little or no changes in the EBA phenotype compared with the respective wild-type control mice. In summary, this suggests, that complement activation in EBA leads to generation of a chemotactic gradient, which supports recruitment of effector cells into the skin, rather than direct toxic effects. This assumption is supported by the observation that autoantibody-induced tissue injury ex vivo, induced by the incubation of cryosections from human skin with anti-COL7 antibodies and neutrophils, is independent of complement activation.
Neutrophils are Key Effector Cells in the Pathogenesis of EBA
Complement activation, and presumably also activation of additional resident immune cells (eg, mast cells and dendritic cells), generates a proinflammatory environment in the skin, leading to the recruitment of leukocytes. If neutrophils are depleted by injection of anti–Gr-1 antibody (clone RB6–8C5) during the induction of EBA by repetitive injections of anti-COL7 IgG, mice are completely protected from disease induction, which strongly indicates that neutrophils are the main effector cells for blister formation and tissue injury. However, a contribution of additional cells, especially monocytes, cannot be excluded, because the Gr-1 antibody is also known to deplete monocytes. The observed resistance of CD18-deficient mice, lacking expression of all β2-integrins, required for firm adhesion and subsequent extravasation of leukocytes to the skin, underscores the significant contribution of neutrophils (and possibly monocytes) in the pathogenesis of experimental EBA.
Neutrophil Activation Leads to the Release of Reactive Oxygen Species and Proteolytic Enzymes, Which Induce Blister Formation
Release of reactive oxygen species (ROS) and proteolytic enzymes is a key event in neutrophil activation. Indeed, neutrophils produce ROS and release their granules upon incubation with immune complexes. In addition, after incubation of cryosections of human skin with anti-COL7 antibodies and neutrophils, formazan precipitates are present at the dermal-epidermal junction when nitroblue tetrazolium is present, indicating ROS production. In contrast, neither formazan precipitates nor dermal-epidermal separation was observed when neutrophils from patients with chronic granulomatous disease, in which the ability of phagocytes to produce ROS is lacking, were used. The dependency of autoantibody-induced tissue injury on ROS production is further supported by the observation of a complete protection of Ncf1-deficient mice, which are unable to produce ROS, from experimental EBA.
In addition to ROS release, neutrophil-derived proteases are indispensable for autoantibody-induced tissue injury in experimental models of EBA. In detail, when cryosections of human skin were treated with autoantibodies from patients with EBA and with peripheral blood leukocytes from healthy donors in the presence of a cocktail of broad-spectrum protease inhibitors, including α2-macroglobulin, α1-protease inhibitor, AEBSF, GM6001, E64, and pepstatin A, leukocytes were recruited to the dermal-epidermal junction but failed to induce split formation at the dermal-epidermal junction. When characterizing the proteases involved more specifically, selective inhibition of human leukocyte elastase or gelatinase B/matrix metalloproteinase-9 (MMP-9) was found to result in the suppression of blistering. Therefore, elastase and gelatinase B/MMP-9 mediate dermal-epidermal separation in the ex vivo models of EBA.
Possible Contribution of Cytokines
There is ample evidence for increased expression of several cytokines in patients with autoimmune blistering skin diseases. However, with few exceptions, there is little experimental evidence that demonstrates a direct contribution of a given cytokine to the pathogenesis of the disease. Regarding EBA, no data on cytokine expression are available. Hence, the contribution of cytokines to the pathogenesis of EBA (and other autoimmune blistering skin diseases) remains an issue of ongoing investigation.
Conclusions and Remaining Questions Regarding Autoantibody-Induced Tissue Damage
Blister formation in experimental models of EBA clearly depends on the autoantibodies’ isotype, complement activation, neutrophils, production of ROS, and release of proteolytic enzymes (see Fig. 2 ). Despite these detailed insights into the pathogenesis of tissue injury in EBA, the exact sequence of events leading to blister formation needs to be defined. Understanding this process in detail may help to identify key molecules in the generation of tissue injury, which, in turn, may be targeted by novel (topical) inflammation-modulating compounds. In addition, several questions regarding the pathogenesis of tissue injury in EBA need to be addressed:
- 1.
What is the contribution of cells other than neutrophils, especially mast cells and monocytes/macrophages, in the pathogenesis of EBA?
- 2.
Are cytokines involved in mediating tissue injury? If yes, which specific cytokines?
- 3.
On the molecular level, which Fc receptors mediate tissue injury?
Related posts:
 A Globally Available Internet-Based Patient Survey of Pemphigus Vulgaris: Epidemiology and Disease Characteristics
A Globally Available Internet-Based Patient Survey of Pemphigus Vulgaris: Epidemiology and Disease Characteristics
 Diagnosis and Clinical Features of Pemphigus Foliaceus
Linear IgA Disease: Clinical Presentation, Diagnosis, and Pathogenesis
Pemphigoid Gestationis: Pathogenesis and Clinical Features
Hair Loss in Autoimmune Cutaneous Bullous Disorders
Nail Involvement in Autoimmune Bullous Disorders
Diagnosis and Clinical Features of Pemphigus Foliaceus
Linear IgA Disease: Clinical Presentation, Diagnosis, and Pathogenesis
Pemphigoid Gestationis: Pathogenesis and Clinical Features
Hair Loss in Autoimmune Cutaneous Bullous Disorders
Nail Involvement in Autoimmune Bullous Disorders
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


