Paraneoplastic pemphigus is a rare condition with extremely high rates of mortality. Although its pathogenesis is incompletely understood, its pathologic findings have significant overlap with other autoimmune blistering diseases, such as pemphigus vulgaris, bullous pemphigoid, erythema multiforme and erosive lichen planus. A universally accepted consensus definition is needed to firmly define the condition. This would aid in identification of paraneoplastic pemphigus and the institution of timely and appropriate treatment to avoid rapid patient deterioration as well as recruitment for trials to further examine the pathogenesis and new therapeutic modalities. This article reviews the varied clinical presentations and pathologic characteristics pertaining to paraneoplastic pemphigus.
Paraneoplastic pemphigus (PNP) is a life-threatening autoimmune blistering disease, which is commonly associated with lymphoproliferative neoplasms. It was first described in a case series in 1990 by Anhalt and colleagues and approximately 250 case reports of PNP have been documented since that time. PNP has a wide geographic distribution and equal predominance across both genders with an average age of onset of 51 years. It is characterized by painful mucosal erosions with a polymorphous skin eruption (not always defined by bullae) in association with an occult or verified neoplasm. These neoplasms are typically of a lymphoproliferative type (chronic lymphocytic leukemia, lymphoma, Castleman’s disease, thymoma, and so forth) although case reports have described rare occurrences of the condition in other neoplasms, such as metastatic melanoma. Mortality due to the disease has been reported to be as high as 90%. Response to therapy is highly variable, with an incomplete knowledge of the pathogenesis of the disease making targeted therapies a challenge. This article reviews the varied clinical presentations and pathologic characteristics pertaining to PNP. Management of PNP will be covered in the next issue of this journal.
Presentations of PNP
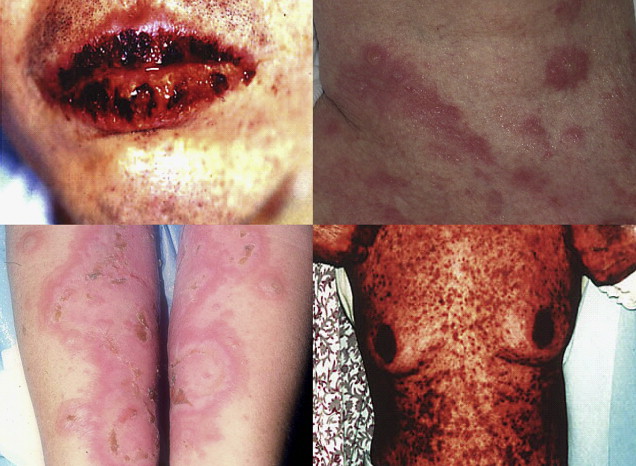
PNP has a variety of clinical presentations, with painful mucosal and skin lesions presenting a wide variation in morphology ( Fig. 1 ). There has also been well-documented evidence of involvement of internal organs, such as lungs, thyroid, kidney, smooth muscle, and gastrointestinal tract. Such internal organ involvement has led to the proposal that the term, paraneoplastic pemphigus , should be replaced by the term, paraneoplastic autoimmune multiorgan syndrome (PAMS ), to more accurately describe the full spectrum of presentations and pathologic findings associated with this condition, although this proposed change has been met with mixed reaction by professionals in the field.
Currently, no consensus definitions exist as to the precise characteristics that define PNP/PAMS. Three different definitions have been put forward by various investigators and are presented in Table 1 . Examples of common aspects present in all proposed definitions of PNP/PAMS include initial mucosal involvement, detection of circulating autoantibodies against envoplakin and/or periplakin, and detection of a neoplasm.
| Proposed Consensus Definitions | Zhu and Zhang | Anhalt et al | Joly et al |
|---|---|---|---|
| Year | 2007 | 1990 | 2000 |
| Clinical Characteristics | Painful mucosal involvement and polymorphic cutaneous eruption | Painful mucosal erosions with a polymorphous skin eruption culminating in vesicles/bullae in the context of an occult/confirmed neoplasm | Presence of painful oral erosions |
| Histopathology | Intraepidermal acatholysis, keratinocyte necrosis, and vacuolar interface dermatitis | Intraepidermal acantholysis, vascular interfacial dermatoses, and keratinocyte necrosis | Suprabasal acantholysis with keratinocyte necrosis or vacuolar interface dermatitis or lichenoid infiltrate |
| DIF | Not specified | Deposition of complement and IgG in intracellular epidermal spaces and in the basement membrane zone in linear granular lesions | Presence of circulating or in vivo bound antiepithelial cell surface and anti–basement membrane zone antibodies |
| IIF | IIF of patient serum with rat bladder epithelia shows intercellular staining | Not specified | Labeling of rat bladder |
| Circulating Autoantibodies | Not specified | Detection in the serum similar to that of pemphigus. | Confirmation of autoantibodies to periplakin and/or envoplakin |
| Presence of a neoplasm | Occult or confirmed | Occult or confirmed | Association with lymphoproliferative disorders |
The clinical presentation of PNP, although highly varied, typically begins with a florid, painful eruption of mucous membrane lesions, which may involve the oropharynx, nasopharynx, tongue, vermillion of the lips, conjunctiva, anogenital region, or esophagus. The onset of cutaneous lesions has a variable delay after the appearance of mucous membrane lesions, ranging from days to months. The heterogeneity in the presentation of the cutaneous manifestations of this disease has led to a stratification of PNP/PAMS based on their similarity to other dermatologic conditions into 5 different clinical variants ( Table 2 ). These cutaneous manifestations often present in waves of lesions and, in contrast to pemphigus vulgaris, blisters in PNP/PAMS do not arise from normal skin but typically develop from inflammatory papules or macules, which greatly outnumber bullous eruptions in PNP/PAMS on individual patients.
| PNP/PAMS Subtype | Pemphigus-like | Bullous Pemphigoid–like | Erythema Multiforme–like | Graft-versus-Host Disease–like | Lichen Planus–like |
|---|---|---|---|---|---|
| Clinical Presentation | Superficial vesicles with occasional erythema | Scaling erythematous papules without bullous lesions | Scaling erythematous papules with occasional ulceration or erosions | Scaled, dusky red papules | Violaceus papules |
| Common Sites of Cutaneous Lesions | Head, trunk, and proximal extremIties | More commonly seen on extremities | Mucosae, trunk, and extremities | Trunk and extremities | Trunk and extremities, which may include acral regions |
| Histologic Findings | Intraepidermal and/or subepidermal bullae | Dyskeratosis and mild mononuclear infiltrate at the dermal-epidermal junction | Dyskeratosis with areas of epidermal separation due to disintegration of the basal layer and perivascular infiltrate | No detachment, hyperparakeratosis, orthoparakeratosis, parakeratosis, and dyskeratosis and pronounced interface dermatitis | No blisters, hyperkeratosis, dyskeratosis, or lichenoid infiltrate |
| DIF Findings | Epidermal intracellular as well as intercellular/linear deposits of IgG and complement C3 | ||||
| IIF Findings | IgG and complement C3 reaction with stratified squamous, transitional and simple columnar epithelium (typically rate bladder and myocardial tissue) | ||||
Related posts:
 A Globally Available Internet-Based Patient Survey of Pemphigus Vulgaris: Epidemiology and Disease Characteristics
A Globally Available Internet-Based Patient Survey of Pemphigus Vulgaris: Epidemiology and Disease Characteristics
 Diagnosis and Clinical Features of Pemphigus Foliaceus
Pemphigoid Gestationis: Pathogenesis and Clinical Features
Pathophysiology of Dermatitis Herpetiformis: A Model for Cutaneous Manifestations of Gastrointestinal Inflammation
Diagnosis and Clinical Features of Pemphigus Foliaceus
Pemphigoid Gestationis: Pathogenesis and Clinical Features
Pathophysiology of Dermatitis Herpetiformis: A Model for Cutaneous Manifestations of Gastrointestinal Inflammation
 Pathogenesis of Epidermolysis Bullosa Acquisita
Nail Involvement in Autoimmune Bullous Disorders
Pathogenesis of Epidermolysis Bullosa Acquisita
Nail Involvement in Autoimmune Bullous Disorders
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


