Panniculitis, Granulomatous, and Fibrosing Disorders
Thomas Stringer
Lori D. Prok
Vikash S. Oza
ERYTHEMA NODOSUM
Definition and Epidemiology
Erythema nodosum (EN), one of the best-recognized panniculitides, is believed to be a delayed-type hypersensitivity reaction in response to a wide variety of stimuli. In the pediatric population, EN is most common among adolescents. EN in children less than 2 years of age is infrequently encountered. In contrast to the far greater incidence of EN in adult females relative to adult males, no such pattern has been found in children.1
Etiology
Approximately 50% of cases of EN are not associated with an underlying etiology and are, therefore, deemed idiopathic. The most common established cause in children is beta-hemolytic Streptococcus infection. In a recent case series of 35 pediatric EN patients, half of the patients were found to have a streptococcal infection of the pharynx.2 Other infectious etiologies include Mycoplasma pneumonia, Epstein-Barr virus (EBV), mycobacteria, and Coccidioides spp. Systemic inflammatory disorders may also give rise to these lesions including sarcoidosis, inflammatory bowel disorder, Behcet disease, pregnancy, drug reaction, or malignancy.3
Clinical Presentation
Similar to adults, EN within children classically presents as pretibial red, tender nodules measuring 1 to 5 cm in diameter (Figure 5-1). Nodules evolve to adopt a reddish-brown or ecchymotic appearance and may persist for 4 to 8 weeks.1 Less common locations include the thighs, arms, trunk, and face. EN should not suppurate or ulcerate and such findings should prompt an evaluation for an alternative diagnosis. Fever, malaise, and arthralgias may precede the development of EN and accompanying lower extremity edema and pain can lead to difficulty with ambulation. Most cases can be managed with supportive care, including bedrest, leg elevation, and nonsteroidal anti-inflammatory drugs. In cases complicated by severe pain, treatment with potassium iodide or colchicine can hasten resolution. Although recurrent cases of pediatric EN have been documented, recrudescence is uncommon.2
Histologic Findings
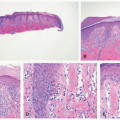
EN is the prototypical septal panniculitis, characterized histologically by a widening of, and inflammation in, the subcutaneous fat septae, with associated septal fibrosis. One often appreciates “spill-over” inflammation into the surrounding fat lobules as well, but low-power microscopic examination usually clearly demonstrates septal involvement as the primary process. The inflammatory cell infiltrate is composed of lymphocytes and histiocytes (Figure 5-2). In acute disease, a varying number of neutrophils and eosinophils are often present, and vasculitis may rarely be noted. Small granulomas formed by the aggregates of histiocytes surrounding a central cleft (so-called Miescher’s radial granulomas) are often present; they are common in, but not pathognomonic for, EN, and can be seen in other forms of panniculitis, as well as in neutrophilic dermatoses and necrobiosis lipoidica (NL) diabeticorum.4,5
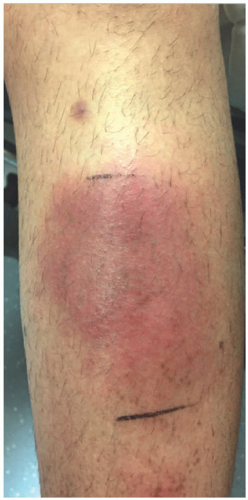 FIGURE 5-1. Erythema nodosum on shin: erythematous, tender plaque on the pretibial shins. |
Differential Diagnosis
In children, EN is, by far, the most common septal panniculitis a pathologist will encounter. Occasionally, the specimen submitted may be suboptimal, or the degree of lobular inflammation may make the identification of the septal-focused process difficult. Chronic EN (subacute nodular migratory panniculitis) demonstrates more impressive septal fibrosis and granulomas. If vasculitis is prominent, thrombophlebitis and cutaneous polyarteritis nodosa should be considered. Occasionally, morphea profunda, deep granuloma annulare (GA), or cutaneous Crohn disease may involve, widen, and/or inflame the fat septae; these entities can usually be distinguished by their characteristic dermal features overlying the foci of panniculitis.
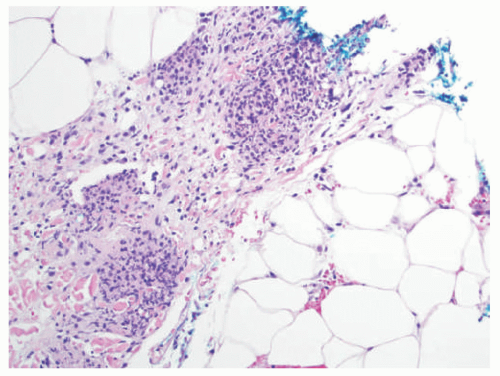 FIGURE 5-2. Erythema nodosum histopathology: widening and chronic inflammation of the subcutaneous fat septae. |
CAPSULE SUMMARY
ERYTHEMA NODOSUM
EN demonstrates a septal (or mixed septal/lobular) panniculitis with a mixed lymphohistiocytic infiltrate, septal fibrosis, and granuloma formation.
ERYTHEMA INDURATUM (NODULAR VASCULITIS)
Definition and Epidemiology
Erythema induratum (EI) was first coined in the mid-19th century to describe an inflammatory panniculitis related to the tuberculosis infection. Since then, identical clinical and histologic patterns have also been documented with other infectious or inflammatory diseases or have been idiopathic. The term “nodular vasculitis” has often been used to describe this EI-like pattern without underlying tuberculosis. Both the clinical and histopathologic presentation of EI and nodular vasculitis are identical. Both EI and nodular vasculitis are exceedingly rare in children and most commonly described in middle-aged adults.
Etiology
Although PCR detection of tuberculosis organisms has substantiated the role of M. tuberculosis in the pathogenesis of EI, the precise etiology is controversial and variably
considered to be a Type III or IV hypersensitivity reaction to bacterial antigen. Nodular vasculitis has been documented with various infections or inflammatory disorders, including sarcoidosis, chronic hepatitis C infection, inflammatory bowel disease, nontuberculosis mycobacterial infections (Mycobacterium chelonae, Mycobacterium avium, and Mycobacterium monacense), and bacteria (Nocardia, Pseudomonas, Fusarium, Chlamydophila pneumonia).6,7,8,9
considered to be a Type III or IV hypersensitivity reaction to bacterial antigen. Nodular vasculitis has been documented with various infections or inflammatory disorders, including sarcoidosis, chronic hepatitis C infection, inflammatory bowel disease, nontuberculosis mycobacterial infections (Mycobacterium chelonae, Mycobacterium avium, and Mycobacterium monacense), and bacteria (Nocardia, Pseudomonas, Fusarium, Chlamydophila pneumonia).6,7,8,9
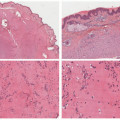
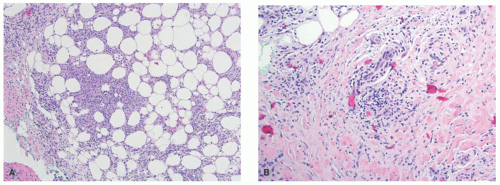 FIGURE 5-3. Erythema induratum histopathology: mixed lobular inflammation (A) and early vasculitis in lobular panniculitis (B). |
Clinical Presentation
EI manifests as symmetrical, tender, deep-seated nodules that may progress to ulceration and heal with atrophic scarring. EI most commonly affects the lower extremities, particularly the calves. Treatment should be carried out expediently with antituberculosis drugs, to which the cutaneous manifestations respond well. Although spontaneous resolution can occur, most cases of EI are chronic and/or recurrent if not treated.
Histologic Findings
EI is histologically identical to nodular vasculitis. Because mycobacteria is not consistently identified in cases of EI, and because numerous inflammatory and other triggers are known to result in the clinical and histologic features of nodular vasculitis, this distinction in nomenclature between the two entities has fallen out of favor within dermatopathology as well, with many authors preferring a unified description.10 EI-nodular vasculitis is characterized histologically by a primarily lobular panniculitis, with lymphohistiocytic granuloma formation and variable necrosis (Figure 5-3A). Vasculitis is the most reliable finding and may present as a necrotizing form in early lesions (Figure 5-3B).11 Careful search for mycobacterial organisms should be done, using special stains (AFB [acid-fast bacilli], FITE) or immunohistochemistry for tuberculosis.
Differential Diagnosis
EI-nodular vasculitis usually involves several adjacent fat lobules, in contrast to polyarteritis nodosa, where the vasculitis involves one large vessel with variable surrounding and associated panniculitis. The other forms of lobular panniculitis are usually excluded by the presence of vasculitis.
CAPSULE SUMMARY
ERYTHEMA INDURATUM (NODULAR VASCULITIS)
EI-nodular vasculitis is a lobular panniculitis with characteristic vasculitis, granuloma formation, and necrosis.
ALPHA-1 ANTITRYPSIN DEFICIENCY PANNICULITIS
Definition and Epidemiology
Alpha-1 antitrypsin deficiency is an inborn error of metabolism leading to variably low blood serum concentrations of the protease inhibitor alpha-1 antitrypsin. Excessive protease activity predisposes patients to chronic obstructive pulmonary disease, hepatic cirrhosis, and panniculitis. Between 80 000 and 100 000 individuals in the United States are thought to suffer from its symptoms.12,13
Etiology
Alpha-1 antitrypsin is an acute-phase reactant that can modulate proteases, most notably neutrophil elastase, as well as the activity of neutrophils, monocytes, macrophages, and mast cells. Its deficiency allows excessive protease activity, leading to tissue damage and the manifestations of the disease. The genes that encode the protein are inherited in
autosomal codominant fashion. The most common deficiency alleles are termed S or Z, whereas the normal allele is designated M. Broadly speaking, MS heterozygotes and SS homozygotes are at low risk for clinically significant disease, MZ and SZ heterozygotes are at greater risk, and ZZ homozygotes develop the most severe symptoms.14
autosomal codominant fashion. The most common deficiency alleles are termed S or Z, whereas the normal allele is designated M. Broadly speaking, MS heterozygotes and SS homozygotes are at low risk for clinically significant disease, MZ and SZ heterozygotes are at greater risk, and ZZ homozygotes develop the most severe symptoms.14
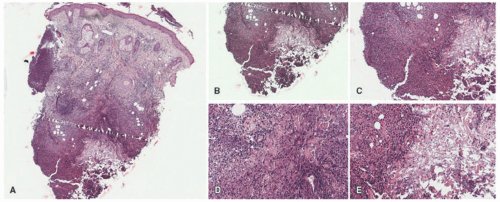 FIGURE 5-4. Alpha-1 antitrypsin deficiency panniculitis. There is mostly lobular panniculitis (A). Prominent liquefactive and subcutaneous necrosis is present. The infiltrate is composed of lymphocytes and histiocytes (B-E). Digital slides courtesy of Path Presenter.com. |
Clinical Presentation
In both pediatric and adult patients, alpha-1 antitrypsin deficiency can present with panniculitis manifesting as purple, tender nodules or plaques, most commonly on the lower trunk, buttocks, and proximal extremities. Lesions can be precipitated by minor trauma and therefore may be misdiagnosed as factitial or artifactual panniculitis. The panniculitis can progress to develop deep, necrotic ulcers that exude oily material. No specific treatment is indicated for the cutaneous findings of this condition—the amelioration of the process with enzyme replacement therapy is sufficient to effect its resolution.
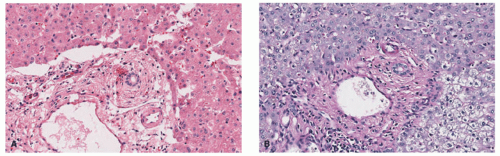 FIGURE 5-5. Alpha-1 antitrypsin deficiency liver abnormalities. A characteristic finding is the presence of periportal globules within the hepatocytes, demonstrated on hematoxylin and eosin (A) and periodic acid-Schiff (B) stains. |
The most common presenting symptom of alpha-1 antitrypsin deficiency in pediatric patients is intrahepatic cholestasis, which in rare cases results in cirrhosis requiring liver transplantation.14 The other extracutaneous manifestations include pulmonary emphysema, pancreatitis, pulmonary embolism or effusion, angioedema, arthritis, or vasculitis.14
Histologic Findings
Alpha-1 antitrypsin deficiency presents as a lobular panniculitis, classically neutrophil-predominant in early lesions, and demonstrating lymphocytes and histiocytes in more long-standing nodules. Liquefactive necrosis of dermal collagen and lipocytes is characteristic, and correlates clinically with oily drainage from ulcerated plaques (Figure 5-4).15 Collagenolysis of fat septae is also common. In the liver, cirrhosis is typical, with the presence of globular inclusions in the periportal areas (Figure 5-5).
Differential Diagnosis
Alpha-1 antitrypsin deficiency is a rare form of panniculitis in childhood. The more common forms of neutrophilic panniculitis should be excluded in the evaluation, including infection, Sweet syndrome and other neutrophilic dermatoses, and foreign objects or injected substances (eg, factitial disorder).16 Special stains for microorganisms and polarized light examination are recommended when evaluating neutrophilic panniculitis. BRAF-inhibitors are becoming more widely used in the pediatric age group for brain and other solid organ tumors and have been associated with drug-induced neutrophilic panniculitis.17,18 This type of panniculitis is not associated with liquefactive necrosis.
CAPSULE SUMMARY
ALPHA-1 ANTITRYPSIN DEFICIENCY PANNICULITIS
Neutrophilic panniculitis in childhood should prompt a consideration of alpha-1 antitrypsin deficiency, although infection is a more common cause of lobular neutrophilic panniculitis, particularly in hospitalized or immunocompromised patients. A medication reaction should also be considered.
LIPOATROPHIC PANNICULITIS
Definition and Epidemiology
Known variously as annular lipoatrophic panniculitis and lipophagic panniculitis, lipoatrophic panniculitis is loosely defined as a panniculitis that heals with fat atrophy accompanied by constitutional symptoms. This entity presents most often in infants and young children 3 to 13 years of age.19
Etiology
The etiology of lipoatrophic panniculitis is poorly understood, although it is likely autoinflammatory in nature, given that cases have been reported with Hashimoto’s thyroiditis, Graves’ disease, juvenile rheumatoid arthritis, and alopecia areata.19
Clinical Presentation
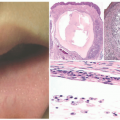
Lipoatrophic panniculitis is characterized by the episodic development of inflamed subcutaneous nodules that expand radially and are followed by permanent lipoatrophy, which is often circumferential around the ankle (Figure 5-6). Lipoatrophic panniculitis follows a relapsing course, with multiple bouts of inflammation and atrophic resolution occurring over the course of months.20 Lesions most often occur on the extremities, and their onset may be preceded by an upper respiratory infection. The clinical differential diagnosis includes deep GA, deep erythema annulare centrifugum, EN, lupus panniculitis, erythema migrans, and morphea. Systemic symptoms associated with this disease include constitutional symptoms, particularly fever, hepatosplenomegaly, arthralgias, and abdominal pain.20,21
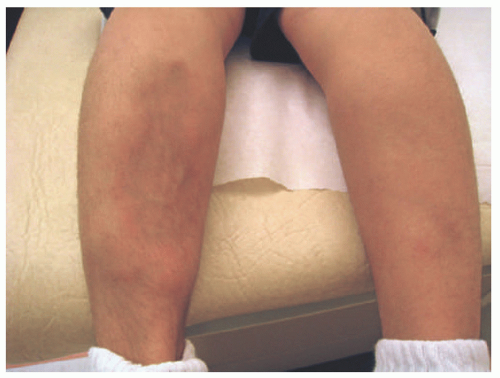 FIGURE 5-6. Lipoatrophic panniculitis: large annular plaque on the medial shin with central atrophy and peripheral erythema and induration. |
Histologic Findings
In early lesions, this disorder presents with lipophagic panniculitis, characterized by a dense mixed inflammatory infiltrate centered on the subcutaneous fat lobules, with numerous foamy lipophages (Figure 5-7A and B).22 Neutrophils are often present in early disease, with histiocytes predominating in later lesions. There is no vasculitis. In very established lesions where clinical atrophy is longstanding, the infiltrate may be sparse, with notable atrophy and fibrosis of the fat lobules.20
Differential Diagnosis
Infection and trauma may both demonstrate lipophagic changes and should be excluded in the evaluation of lipoatrophic panniculitis. The various forms of neutrophilic panniculitis may be considered in early lesions. Once atrophy predominates, the findings may not be distinguishable from other forms of lipoatrophy and lipodystrophy; these disorders are usually able to be excluded on the basis of clinical history. Several reports have described lymphocytes rimming adipocytes as well as mildly atypical lymphocytes infiltrating subcutaneous vessels; these findings may prompt a consideration of subcutaneous panniculitis-like T-cell lymphoma, which can be excluded by the lack of necrosis or lymphocyte phagocytosis by histiocytes.23
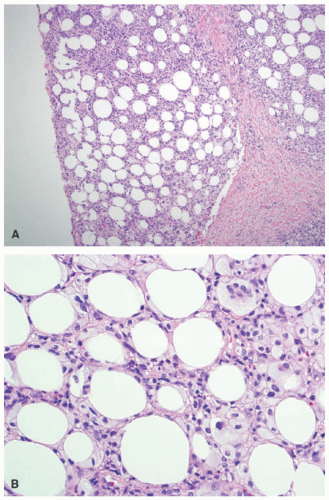 FIGURE 5-7. Lipoatrophic panniculitis histopathology: lobular panniculitis with fat necrosis (A). High-power view highlighting lipophagic fat necrosis and foamy histiocytes (B). |
CAPSULE SUMMARY
LIPOATROPHIC PANNICULITIS
Lipoatrophic or lipophagic panniculitis presents histologically with a mixed lobular infiltrate, with lipid-laden macrophages, and lobular atrophy, likely triggered by an autoimmune or inflammatory process.
COLD-INDUCED PANNICULITIS
Definition and Epidemiology
Cold-induced panniculitis is an eruption of subcutaneous nodules in response to prolonged cold exposure. Although cold panniculitis may present well into adulthood, most cases, including the initial reports, tend to occur in young children.24
Etiology
The predilection of cold-induced panniculitis for younger patients is thought to arise because of a higher ratio of unsaturated to saturated fatty acids in the subcutaneous fat, the former of which have a higher freezing point.
Clinical Presentation
Cold-induced panniculitis presents with painful, poorly defined erythematous plaques or subcutaneous nodules (Figure 5-8). The lesions often persist for hours to days post exposure and resolve spontaneously. The sources of exposure include cold ambient temperatures, ice packs, or cooling blankets.25 The phenomenon known as “popsicle panniculitis” is a well-described presentation in teething children given frozen substances as an analgesic and the subsequent development of panniculitis involving the cheeks or submental area.26
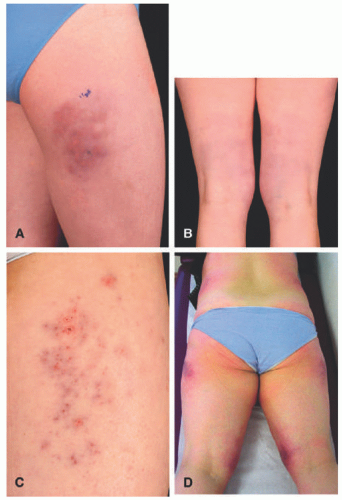 FIGURE 5-8. Cold panniculitis. Clinical aspects of cold-associated perniosis of the thighs. A, Typical presentation with livid-red, infiltrated lesions on the lateral aspect of the thigh; B, similar clinical presentation but lesions located on the posterior aspect of both thighs; C, partly eroded lesions located on the lateral aspect of the thigh; D, widespread involvement with lesions located on the lateral and medial aspects of both thighs as well as both flanks. Reprinted with permission from Ferrara G, Cerroni L. Cold-associated perniosis of the thighs (“equestrian-type” chilblain): a reappraisal based on a clinicopathologic and immunohistochemical study of 6 cases. Am J Dermatopathol. 2016;38(10):726-731. |
Histologic Findings
A histologic hallmark of cold panniculitis is the presence of edema in the papillary dermis; as such, incisional specimens that do not include overlying superficial dermis are not ideal when evaluating for this entity. Cold panniculitis presents with mixed acute and chronic lobular inflammation, with characteristic adipocyte necrosis (Figures 5-9 and 5-10). A clinical history of cold exposure is helpful in evaluation.27
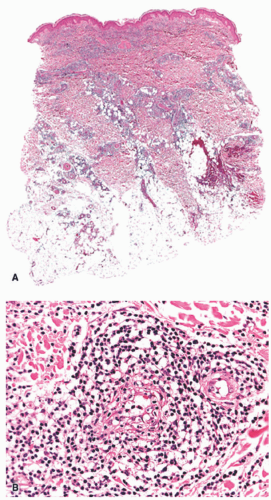 FIGURE 5-9. Cold-associated panniculitis of the thighs. A, Dense inflammatory infiltrates predominantly located within the dermis and only superficial portion of subcutaneous fat. B, Prominent involvement of a vessel with features of lymphocytic venulitis. Reprinted with permission from Ferrara G, Cerroni L. Cold-associated perniosis of the thighs (“equestrian-type” chilblain): a reappraisal based on a clinicopathologic and immunohistochemical study of 6 cases. Am J Dermatopathol. 2016;38(10):726-731. |
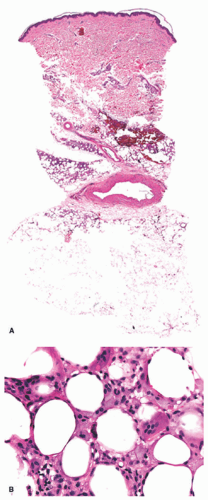 FIGURE 5-10. Cold-associated panniculitis. Late stage of cold-associated perniosis of the thighs with lipophagic features in the upper part of the subcutaneous fat (B); only minimal dermal and subcutaneous inflammatory infiltrate present (A). Reprinted with permission from Ferrara G, Cerroni L. Cold-associated perniosis of the thighs (“equestrian-type” chilblain): a reappraisal based on a clinicopathologic and immunohistochemical study of 6 cases. Am J Dermatopathol. 2016;38(10):726-731. |
Differential Diagnosis
Other “physical” causes of panniculitis can show similar histologic features to cold panniculitis, including traumatic and factitial panniculitis. All can present with acute inflammation and necrosis. Clinical-pathologic correlation is required in most cases. Polarized light examination should be performed to exclude foreign material.
CAPSULE SUMMARY
COLD-INDUCED PANNICULITIS
Cold panniculitis demonstrates a lobular panniculitis with mixed inflammation and marked papillary dermal edema. Clinical-pathologic correlation is helpful in making the correct diagnosis.
LUPUS PANNICULITIS
Definition and Epidemiology
Lupus erythematosus panniculitis (LEP) represents a subcutaneous variant of cutaneous lupus erythematosus. LEP is an uncommon diagnosis in children, believed to represent only 2% of pediatric patients with cutaneous systemic lupus erythematosus (SLE). The mean age of onset of LEP is 8 years.28
Etiology
Clinical Presentation
LEP often presents with firm, asymptomatic, sharply demarcated, rubbery dermal plaques or nodules most commonly on the face and/or upper arms in children.30 As inflammation subsides, lipoatrophy typically develops, placing patients at risk for permanent disfigurement. LEP should be distinguished from EN, another common form of panniculitis in patients with SLE. Unlike EN, LEP does not typically involve the shins, is chronic in duration, and is typically painless. The diagnosis of LEP is best established by a skin biopsy, and laboratory workup is infrequently helpful. A positive antinuclear antibody (ANA) is inconsistently found in children with LEP.30
Histologic Findings
Lupus panniculitis is characterized by dense lobules of lymphocytes often centered at the periphery of the fat lobules in association with marked fibrosis. A mixed lobular and septal panniculitis may be seen. Histiocytes and plasma cells are usually also present. Lymphoid follicles are typical. There is reliable hyaline necrosis of the fat lobules, with associated edema, mucin deposition, and variable calcification (Figure 5-11).5,31 The overlying epidermis frequently demonstrates classic interface changes of lupus, with basal vacuolar change, dyskeratosis, and lymphocytic inflammation at the dermo-epidermal junction and follicles.32
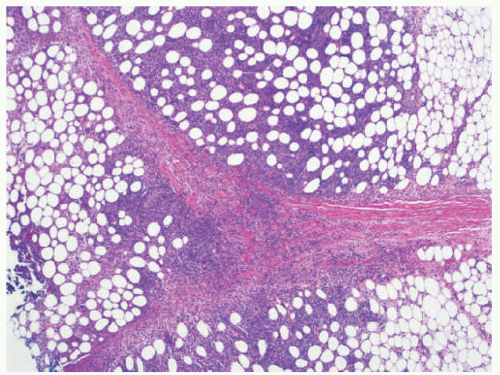 FIGURE 5-11. Lupus panniculitis: dense lymphocytic lobular panniculitis and hyalinized fat septae. |
Differential Diagnosis
Panniculitis associated with dermatomyositis demonstrates identical histologic features to lupus panniculitis and must be distinguished clinically. The other forms of lobular panniculitis can be excluded on the basis of hyaline necrosis, mucin deposition, and overlying epidermal and papillary dermal changes.
CAPSULE SUMMARY
LUPUS PANNICULITIS
Lupus panniculitis demonstrates dense mixed but lymphocyte-predominant lobular panniculitis with hyaline fat necrosis. The overlying epidermis and papillary dermis should always be examined for evidence of interface changes and lichenoid inflammation.
SUBCUTANEOUS PANNICULITIS-LIKE T-CELL LYMPHOMA
Definition and Epidemiology
Subcutaneous panniculitis-like T-cell lymphoma (SPTCL) is a panniculitis of neoplastic origin driven by cytotoxic T-cells with the α/β lineage.33 The former cases with a γ/δ phenotype are now designated as primary cutaneous γ/δ
T-cell lymphoma. SPTCL may present in a broad age range. A review of 83 cases found a mean age of 36, with a range extending from 9 to 79 years of age.34
T-cell lymphoma. SPTCL may present in a broad age range. A review of 83 cases found a mean age of 36, with a range extending from 9 to 79 years of age.34
Etiology
It is unclear whether or to what extent SPTCL is a reactive T-cell lymphoproliferative syndrome, as opposed to a primary malignancy in its own right. SPTCL is well described as occurring in patients previously diagnosed with LEP, and there is a striking histopathologic similarity between the two.35,36
Clinical Presentation
SPTCL presents in similar fashion to LEP, rendering the distinction between the two on clinical grounds difficult, although case series indicate that the latter disease often possesses more prominent constitutional symptoms, leukopenia, and increased erythrocyte sedimentation rate.36 The cases with associated hemophagocytosis have a more aggressive clinical course.
Much ambiguity exists regarding the clinical separation of SPTCL, LEP, and the entity cytophagic histiocytic panniculitis (CHP), the latter an idiopathic febrile panniculitis in the setting of hemophagocytosis, also considered to represent a reactive lymphocytic process. The symptomatology of these disorders, which often includes constitutional symptoms, cytopenias, hepatosplenomegaly, and other end organ involvement, makes them difficult to distinguish. Moreover, it is an open question whether or not CHP and SPTCL fall onto a continuum of neoplastic disease or whether they represent their own separate disorders with unique clinical spectra of presentation.37,38,39
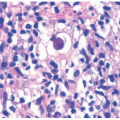
Histologic Findings
A dense, monotonous infiltrate of small- to medium-sized atypical lymphocytes infiltrate the subcutaneous fat lobules, giving the appearance of a lobular panniculitis. Tumor cells demonstrate karyorrhexis and pleomorphism. Mixed inflammation and granulomatous foci may be present, and fat necrosis is common. “Rimming” of individual adipocytes by tumor cells is a characteristic feature, although not specific.40 Vascular invasion is variably present. Immunohistochemistry highlights cytotoxic T-cells, with a CD8 predominant phenotype (CD3+, CD4-, CD8+, CD56-). All SPTCL are BF1+ (TCR alpha/beta phenotype). Ki67 shows frequent increased proliferation among the neoplastic cells.
Differential Diagnosis
SPTCL may be mistaken for a benign or reactive lobular panniculitis, particularly lupus panniculitis. Cytologic atypia, rimming of adipocytes, lack of any dermal involvement, and the absence of interface changes in the overlying epidermis help distinguish SPTCL. LEP typically has pronounced fibrosis and lymphoid follicles. Other lymphomas may be excluded on the basis of immunohistochemistry.
CAPSULE SUMMARY
SUBCUTANEOUS PANNICULITIS-LIKE T-CELL LYMPHOMA
SPTCL presents histologically as an atypical lymphocytic infiltrate confined to the subcutis, mimicking a lobular panniculitis.
NEUTROPHILIC LOBULAR PANNICULITIS
Definition and Epidemiology
Neutrophilic lobular panniculitis is a neutrophil-driven, principally lobular panniculitis often presenting alongside relapsing fevers and other constitutional symptoms. Neutrophilic lobular panniculitis is best described as a histopathologic reaction pattern and not as a distinct clinical entity, which can be associated with various cutaneous and systemic disorders.
Etiology
Infection, Sweet syndrome and other neutrophilic dermatoses, foreign objects or injected substances, trauma, early lipophagic panniculitis, alpha-1 antitrypsin deficiency, and autoimmune diseases (particularly rheumatoid arthritis and inflammatory bowel disease) may all be associated with neutrophilic panniculitis.16 A special consideration in the pediatric age group is neutrophilic panniculitis as a reaction to BRAF inhibitor medications, the use of which are becoming more frequent in the treatment of cutaneous and solid organ malignancies.18
Clinical Presentation
Any of the aforementioned etiologies may manifest with subcutaneous nodules in concert with recurrent fevers. Patients with this clinical presentation and histologic features of a neutrophilic panniculitis must undergo a thorough infectious disease workup.
The term Weber-Christian disease, which denotes a febrile panniculitis of unknown origin, has fallen out of favor in the dermatologic literature following a 1998 study in which alternative clinicopathologic diagnoses were found in all 30 examined adult cases previously diagnosed as Weber-Christian. It is now considered a placeholder for the panniculitis of etiology yet to be uncovered.41
Histologic Findings
As the name indicates, neutrophilic panniculitis is characterized by a neutrophil-predominant inflammatory cell
infiltrate of the fat lobules with a relative sparing of the septi (Figure 5-12). Leukocytoclasis can be very prominent in the absence of frank vasculitis.
infiltrate of the fat lobules with a relative sparing of the septi (Figure 5-12). Leukocytoclasis can be very prominent in the absence of frank vasculitis.
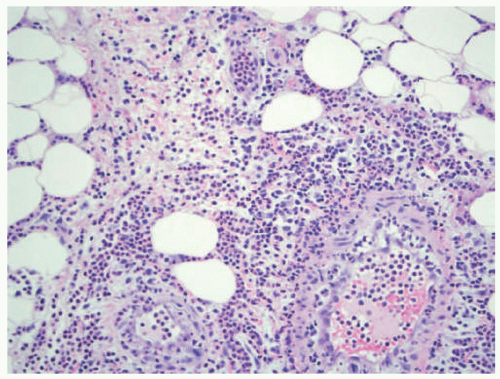 FIGURE 5-12. Neutrophilic lobular panniculitis histopathology: neutrophil-predominant lobular infiltrate in a patient treated with BRAF inhibitor therapy for glioblastoma. |
Differential Diagnosis
Some cases of neutrophilic panniculitis may demonstrate variable septal inflammation. In these cases, EN and Sweet syndrome involving the subcutis should be excluded. Special stains for microorganisms and polarized light examination are always recommended when evaluating neutrophilic panniculitis.
CAPSULE SUMMARY
NEUTROPHILIC LOBULAR PANNICULITIS
Neutrophilic panniculitis is a reaction pattern characterized by neutrophilic lobular or mixed lobular and septal inflammation. Medication reaction to BRAF inhibitor is a well-documented cause of neutrophilic panniculitis in children. Several other cutaneous and systemic disorders can present with similar histologic features.
TRAUMATIC/FACTITIAL PANNICULITIS
Definition and Epidemiology
Factitial, or traumatic, panniculitides are produced by adventitious injury. Factitial panniculitides are most often described in young adults and middle-aged women.
Etiology
The causes of these injuries are commonly grouped into mechanical (eg, traumatic), physical (eg, arising from application of cold or heat), and chemical. It is important to note that panniculitides arising from iatrogenic sources are considered to be factitial. The most salient agents for pediatric purposes are vaccination, particularly antihepatitis vaccination or antitetanus toxoid, and phytonadione, or vitamin K. A subcutaneous reaction pattern can be induced by penetrating or blunt trauma, the injection of foreign substances, or persistent local pressure. Surreptitiously injected substances may include mineral or vegetable oils, which induce subcutaneous inflammation.
Clinical Presentation
The morphology of the lesions may vary depending upon the nature of the insult. Panniculitis ensuing from penetrating trauma may be well demarcated and/or sharply angulated, whereas nodules induced by blunt trauma may appear ecchymotic, often arising on the arm or hand. The diagnosis is made more difficult by the idiosyncratic reactions of injected agents, such as Texier disease in the context of vitamin K injections. Early in the course, these may appear as robust eczematous reactions, whereas late presentations may resemble sclerotic lesions with a lilac border. Careful evaluation of the clinical history and distribution must be undertaken to arrive at the proper diagnosis.
Histologic Features
Acute or chronic trauma of the fat presents histologically as a lobular panniculitis, with chronic mixed inflammation and foci of fat necrosis and lipomembranous change. Low-power examination often reveals cystic spaces in the fat lobules. Hemorrhage and hemosiderosis are common, and fibrosis is usually present in chronic lesions. Clinical hypertrichosis has been reported overlying plaques of traumatic panniculitis, but is not associated with histologic changes of the hair follicles.42 A “mobile encapsulated lipoma” is a discrete focus of traumatized and necrotic fat that has been isolated in the skin (Figure 5-13A). It is often surprisingly mobile beneath the skin surface. It demonstrates identical histologic features, but in a small, encapsulated deep dermal nodule (Figure 5-13B).43
Differential Diagnosis
Cold panniculitis, foreign material, and other forms of factitial panniculitis may all present with mixed lobular inflammation and fat necrosis. Clinical history is critical in distinguishing these entities. Histologic features that may be helpful include dermal edema (frequent in cold panniculitis), foreign material demonstrated on polarized light examination, and the presence of irregular “Swiss cheese-like” cystic spaces in forms of factitial panniculitis.
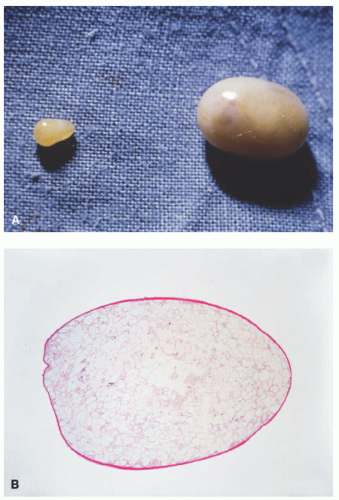 FIGURE 5-13. A, Mobile encapsulated lipoma gross: shiny, encapsulated, soft nodule; easily excised intact. B, Mobile encapsulated lipoma histopathology: well-circumscribed and encapsulated aggregate of necrotic lipocytes. |
CAPSULE SUMMARY
TRAUMATIC/FACTITIAL PANNICULITIS
Traumatic panniculitis demonstrates mixed lobular in-flammation with necrosis, hemorrhage, and fibrosis.
GRANULOMA ANNULARE
Definition and Epidemiology
GA is a noninfectious granulomatous skin condition presenting with variable clinical morphologies. In general, GA is most common in school-aged children. The subcutaneous variant preferentially occurs in young children, with the most number of children being less than 6 years of age.
Etiology
GA is believed to be caused by a delayed-type hypersensitivity reaction due to unknown stimulating antigens.44 Most cases of childhood GA occur in the absence of an underlying medical condition. Risk factors associated with GA include diabetes mellitus, tetanus or BCG vaccination, Hepatitis B viral infection, Borrelia infection, Hodgkin’s lymphoma, and acute lymphocytic leukemia.25
Clinical Presentation
The lesions of GA are comprised of small skin colored to pink/violaceous individual papules coalescing to form an annular or circinate plaque without scale (Figure 5-14). The four most common clinical variants are localized, generalized, subcutaneous, and perforating GA. Localized GA typically involves the extremities, specifically the wrists, ankles, and the dorsal surface of the hands and feet. By definition, generalized GA must involve the trunk and at least one set of extremities and may follow a more protracted course than other variants.45 The perforating morphology may present as grouped papules with central umbilication, crusting, and scaling. Subcutaneous GA presents with subcutaneous nodules on the feet, anterior tibial surface, fingers, hands, or scalp. This variant can appear clinically similar to rheumatoid or pseudorheumatoid nodules, but both of these diagnoses are much less common in children than in adults.46
GA is a self-limited disorder with 50% of lesions resolving in 2 years, and therefore, observation is a reasonable management approach.47 Potent topical corticosteroids, intralesional corticosteroids, and cryotherapy have all been used to treat individual lesions. Occasionally, severe and generalized disease may require systemic medication, and isotretinoin, antimalarial (plaquenil), and dapsone have been used in this setting.25
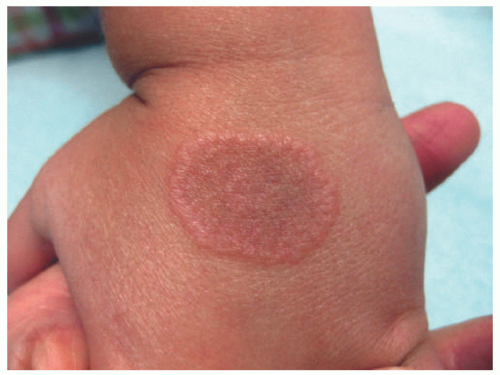 FIGURE 5-14. Granuloma annulare: erythematous, annular plaque with a raised border of monomorphous papules. Courtesy of Anna Bruckner, MD. |
Histologic Findings
Classic GA is characterized histologically by necrobiotic granulomas in the superficial and mid dermis. These demonstrate altered, eosinophilic, and thickened collagen at the center, with lymphocytes and histiocytes surrounding in a palisaded manner (Figure 5-15). Eosinophils may be present, and scattered multinucleate histiocytes are frequent. Mucin deposition is present in the necrobiotic foci, a feature that helps distinguish GA from other granulomatous infiltrates. The mucin can be demonstrated with the use of Alcian blue or colloidal iron stains. A perivascular lymphocytic infiltrate is usually noted in the surrounding dermis. In the interstitial form of GA, histiocytes intercalate between dermal collagen bundles, giving a “busy” appearance to the dermis at low-power examination, without distinct necrobiotic foci. Interstitial mucin is present.
Differential Diagnosis
NL diabeticorum can appear similar to GA histologically, but generally shows pandermal granulomas rather than discrete foci and a lack of mucin deposition. Rheumatoid nodules are larger, demonstrate more fiery pink collagen with fibrin deposition and necrosis, and are present deeper in the dermis. The presence of numerous multinucleate histiocytes and/or acute inflammation should prompt the consideration of a foreign body granuloma and the performance of polarized light examination. Infection should also be considered if granulomas and neutrophilic inflammation are present. Occasionally, the presence of dense and well-formed granulomas without definitive necrobiosis may mimic features of cutaneous sarcoidosis.48 The interstitial form of GA demonstrates histologic features similar to an interstitial drug eruption and some forms of scleromyxedema.49,50 If neutrophils are present in association with interstitial granulomatous inflammation, “palisaded neutrophilic and granulomatous dermatitis” (which may be associated with a variety of underlying diseases including rheumatoid arthritis, lupus and other connective tissue disorders, inflammatory bowel disease, myeloproliferative disorders, infection, and medications) should be considered.51,52
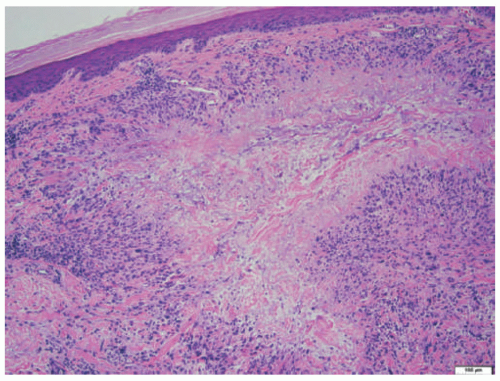 FIGURE 5-15. Granuloma annulare histopathology: necrobiotic granuloma, demonstrating central eosinophilic collagen and mucin, with surrounding palisaded lymphohistiocytic inflammation.
Related posts:Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|