Classification
Percent of pediatric cases (%)
Description
Associated signs and symptoms
Prognosis
Cutaneous solitary mastocytoma
20
Solitary macule or plaque with mast cell infiltrate
Itching, lymphadenopathy
Excellent: >90% show partial or complete regression
Urticaria pigmentosa/maculopapular cutaneous mastocytosis
75
Multiple macules, papules, plaques, or bullae
Itching, flushing, diarrhea, dyspnea, hepatomegaly lymphadenopathy, splenomegaly
Very good: 71% show partial or complete regression
Diffuse cutaneous mast ocytosis
5
Diffuse mast cell infiltration of skin, often causing peau d’orange appearance; may have overlying papules or vesicles
Itching, hepatomegaly, lymphadenopathy, splenomegaly, diarrhea
Excellent: >90% show partial or complete regression
Telangiectasia macularis eruptiva perstans ( TMEP)
<1
Brown macules, often on trunk. Scant mast cell infiltrate, often with negative Darier’s sign
Limited data in children, but often asymptomatic. About half of adults have associated systemic mastocytosis
Long-term prognosis unknown, reported cases have benign course
Epidemiology
There are no epidemiologic studies of pediatric cutaneous mastocytosis. Overall estimates of prevalence of mastocytosis in European populations are around 9–13 per 100,000 [6]. Pediatric mastocytosis is largely a benign condition, with about 75% of children reported in recent literature presenting with urticaria pigmentosa, 20% with solitary mastocytoma, 5% with diffuse cutaneous mastocytosis, and <1% with TMEP [3]. Systemic and malignancy-related forms are rarely encountered in pediatric populations.
Natural History
About 90% of cases of cutaneous mastocytosis present prior to age two, with a slight male predominance [2, 3]. In children with early-onset disease, lesions tend to resolve by adolescence [7]. One case series, which followed 13 patients with UP and 2 patients with DCM for over 20 years, demonstrated that 10 of 15 showed complete resolution, and the remaining 5 showed partial or major improvement [8]. Pediatric patients with small lesions (<1 cm) show significantly later resolution that those with larger lesions, higher tryptase, and later onset of disease [9]. Across all types of pediatric mastocytosis, 67% of patients show partial or complete regression [3].
Though the majority of patient’s signs and symptoms resolve or gradually improve, a smaller subset may have progressive disease. In a review of published cases, Meni et al. 2015 noted that children with UP were the most likely to have cutaneous worsening or develop systemic mastocytosis . In the same review, 2.9% of cases with follow-up data resulted in a fatal outcome; however, this is likely heavily affected by publication bias [3].
Pathogenesis
The cause of mastocytosis is unknown, but genetic factors have been proposed. Hematopoietic stem cells differentiate into mast cells in response to signaling through CD117 , a transmembrane tyrosine kinase receptor [10]. Adults and children with cutaneous mastocytosis have been shown to harbor mutations in c-KIT , the gene encoding CD117, though the affected codon differs in pediatric patients in comparison to adult patients [11, 12]. In adults, KIT mutations are commonly found on exon 17, KIT D816V, whereas children demonstrate mutations in exon 8, 9, or 17 [12]. c-Kit is a receptor tyrosine kinase essential for numerous cellular signal transduction pathways [13]. The significance of genetic polymorphisms in c-Kit among pediatric patients is not yet clear, and the utility of genetic testing in clinical practice has not been established.
Symptoms of mastocytosis are largely due to the activation and degranulation of mast cells, resulting in release of a variety of mediators, including proteases, histamine, proteoglycans, cytokines, prostaglandins, and leukotrienes [14]. Histamine release from mast cells can lead to symptoms in all forms of cutaneous mastocytosis (Table 4.2), ranging from mild headache, nausea, vomiting, diarrhea, and cutaneous flushing to severe symptoms such as hypotension, anaphylaxis, and neurologic dysfunction [15].
Skin symptoms |
Itching |
Flushing |
Gastrointestinal symptoms |
Diarrhea |
Abdominal pain |
Vomiting |
Respiratory symptoms |
Wheezing/dyspnea |
Cough |
Rhinorrhea |
Other |
Bone pain |
Headache |
Systemic symptoms |
Irritability |
Hypotension |
Anaphylaxis |
Syncope |
Clinical Features
Mastocytosis has a variety of clinical manifestations from limited skin involvement to systemic signs and symptoms. The rate of anaphylaxis in children with all types of mastocytosis is estimated to be 5–9% [3, 17]. Rates in urticaria pigmentosa are lower, around 1.5%, but greater cutaneous involvement is associated with higher risk of anaphylaxis [16, 17]. In one study, 13% of adults with cutaneous mastocytosis reported a history of anaphylaxis, whereas in adults with systemic mastocytosis rates of anaphylaxis have been reported to be as high as 56% [17].
Clinical manifestations may vary by mastocytosis subtypes. Pediatric cutaneous mastocytosis subtypes include solitary mastocytoma (SM) , maculopapular cutaneous mastocytosis or urticaria pigmentosa (UP) , diffuse cutaneous mastocytosis (DCM) , and telangiectasia macularis eruptiva perstans (TMEP), with UP representing the most common variant (see Table 4.1). Rare cases of localized xanthelasmoid mastocytosis have been reported, often favoring flexural areas, with appearance similar to pseudoxanthoma elasticum or juvenile xanthogranuloma [18–20].
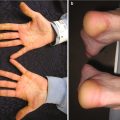
About 20% of cases of pediatric mastocytosis present as a solitary mastocytoma [3]. Solitary mastocytomas typically present in early childhood as a single yellow-brown lesion. Induration of skin with associated dimpling resulting from cutaneous infiltration has been referred to as having a peau d’orange (or orange skin) appearance (Fig. 4.1). Systemic symptoms are rarely encountered in association with solitary mastocytomas, and >90% of lesions will spontaneously resolve [3].
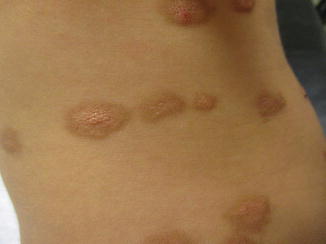
Fig. 4.1
Cutaneous mastocytosis. Several lesions in this patient exhibit peau d’ orange textural changes
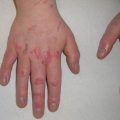
Urticaria pigmentosa presents with multiple yellow-brown macules, papules, plaques, or blisters. Lesions range from a few millimeters to 1–2 cm, tend to first appear on the trunk, and generally spare acral regions. Blistering can be seen, most prominently in association with lesions on the head [21]. Blistering tends to appear and resolve in the first few years of life [22]. In patients with UP, both the number and severity of skin lesions predict systemic symptoms, most commonly gastrointestinal, respiratory, or neuropsychiatric disturbances [16].
Patients with DCM present with generalized hyperpigmentation and induration of skin, as opposed to the discrete lesions seen in UP or SM. Two cutaneous manifestations of DCM have been described, including indurated plaques or blisters with background generalized erythema and yellow/orange plaques mimicking xanthomatous skin lesions [23, 24]. Systemic symptoms such as flushing, GI distress, and bone pain are more common in DCM than other forms of cutaneous mastocytosis [3, 15].
Telangiectasia macularis eruptiva perstans (TMEP) is a form of cutaneous mastocytosis seen in <1% of childhood cutaneous mastocytosis, and in up to 14% of adult cases, with median age of onset at 50 years [5]. TMEP is characterized by small red-brown telangiectatic macules more commonly located on extremities. Up to 50% of adult patients with TMEP experience systemic symptoms [5], but there is a paucity of pediatric data due to the rarity of this presentation in children.
It is important to note the distinctions in clinical features between pediatric and adult-onset mastocytosis. Adult-onset mastocytosis is much more likely to be associated with systemic manifestations, is less likely to regress, and presents with smaller, monomorphic lesions on the trunk and thighs. Children typically have larger, polymorphic macules, papules, plaques, or blisters targeting the head and neck, trunk, and extremities [21]. Interestingly, children with small (<1 cm), monomorphic maculopapular lesions may have a later onset in disease and higher likelihood of disease persistence beyond childhood [9].
Though pediatric fatalities in the perioperative period have not been reported, the perioperative period represents a clinical situation that requires special attention. While 2–4% of children with cutaneous mastocytosis may experience moderate symptoms secondary to mast cell release and/or anaphylaxis from anesthesia, such complications may be avoided with prophylactic antihistamines [25]. Importantly, there are no reported cases of anesthesia-related complications in patients with solitary mastocytoma [26]. The incidence in pediatric systemic mastocytosis is unknown due to rarity of the diagnosis. The perioperative period may introduce numerous triggers, including stress, temperature change, mechanical stress, and medications. Therefore, anesthesiologists should be aware of the patient’s diagnosis and obtain a thorough history of past triggers. Most authors advocate for continued administration of maintenance mastocytosis-directed treatment, incremental administration of medications with potential for mast cell release, and substitution of fentanyl or sufentanil for known degranulators, including atracurium, mivacurium, meperidine, and morphine [26]. NSAIDs may be used, with caution, in children without known sensitivity. Regardless of procedure, medication list, or severity of cutaneous symptoms, medical providers should be prepared to respond to episodes of anaphylaxis in children with cutaneous mastocytosis undergoing anesthesia.
Diagnosis
The diagnosis of cutaneous mastocytosis is established by identifying the highly characteristic clinical exam (see Fig. 4.1) and/or with skin biopsy. Lesions of cutaneous mastocytosis are hyperpigmented, fixed macules, papules or plaques that tend to exhibit a wheal and flare reaction when stroked, known as the Darier’s sign . Skin biopsy of suspected lesions demonstrates a mast cell infiltrate in the papillary dermis. Increase in mast cell number can be subtle, particularly in patients with TMEP . Giemsa, toluidine blue, or immunohistochemical stains with CD117/c-kit and tryptase can help to identify mast cells on skin biopsy.
Many experts suggest that evaluation for systemic mastocytosis is unnecessary for most children with mastocytosis, but for certain patients with severe disease, further workup may be warranted. Though controversial, some authors recommend consideration of systemic workup in the following scenarios: organomegaly, elevated tryptase, unexplained peripheral blood abnormalities, or persistence of cutaneous lesions after puberty. Differentiation of cutaneous mastocytosis from systemic mastocytosis generally requires a bone marrow biopsy and is supported by the criteria listed in Box 4.1 [27].
Box 4.1 Criteria for the diagnosis of systemic mastocytosis, adapted from Valent et al. 2001 [27]
Major
- 1.
>15 mast cells in aggregates on bone marrow biopsy or other extracutaneous tissue
Minor
- 1.
25% spindle-shaped mast cells in extracutaneous specimen or >25% atypical mast cells
- 2.
C-kit mutation in codon 816
- 3.
Co-expression of kit and CD2 and/or CD25 in mast cells in bone marrow, blood, or extracutaneous tissue
- 4.
Serum tryptase persistently >20 ng/mL
One major and one minor or three minor criteria are required for the diagnosis of systemic mastocytosis.
Laboratory Findings
Recommendations for initial laboratory testing vary between authors and clinicians. Some suggest that CBC with differential and biochemistry should be obtained at diagnosis in addition to baseline tryptase in those with UP or DCM [28]. Frequency of repeat testing has not been established but likely should be based on the severity of systemic symptoms.
Monitoring of tryptase levels may be helpful in determining burden of cutaneous disease and risk for systemic involvement. Children with diffuse cutaneous mastocytosis display significantly higher levels than those with urticaria pigmentosa or solitary mastocytoma [15, 29]. Serum tryptase tends to decrease over time and correlate with improvement in symptoms. As such, increases in serum tryptase may indicate the need for further evaluation and consideration of bone marrow biopsy [30]. In a case series of 105 children evaluated for cutaneous mastocytosis, systemic mastocytosis was found only in children with both elevated tryptase (>20 ng/mL) and organomegaly, suggesting that these factors should be considered in the decision to pursue bone marrow biopsy in pediatric patients [30]. Importantly, there were no patients with elevated tryptase in the absence of organomegaly who were found to have systemic mastocytosis [30]. Plasma histamine, though elevated in most cases of CM, has not been found correlate with active disease in mastocytosis [31].
The SCORing MAstocytosis (SCORMA) index correlates with serum tryptase and has been proposed as a standardized method to grade severity of mastocytosis [32]. This index combines the anatomical area involved, intensity of cutaneous findings, and subjective symptoms reported by patients or families.
Treatment
Treatment of mastocytosis is dependent on the severity of symptoms relating to mast cell degranulation and mast cell mediator release (Table 4.3). Treatment can successfully ameliorate symptoms but does not appear to hasten resolution of mastocytosis. A detailed protocol for management is proposed by Heide et al. 2008 [33]. For patients with severe disease, avoidance of triggers of mast cell degranulation (Table 4.4) can help prevent systemic symptoms.
Clinical presentation | Treatment recommendations |
|---|---|
All forms | At least yearly follow-up with abdominal and lymph node exam Screening for systemic symptoms of mast cell degranulation |
Asymptomatic cutaneous lesions | No treatment required |
Cutaneous solitary mastocytoma | Topical steroids may speed partial regression [34] Consider surgical excision if severe |
Urticaria pigmentosa or diffuse cutaneous mastocytosis with symptoms from histamine release (see Table 4.2) | First- and/or second-generation antihistamines Oral cromolyn sulfate Topical steroids Avoid known triggers Consider epinephrine prescription |
Extensive cutaneous disease | Avoid known triggers Provide epinephrine prescription PRN |
Langerhans Cell Histiocytosis
Key Points
Langerhans cell histiocytosis (LCH) is a neoplastic disorder arising from histiocytes that can affect nearly any organ system.
Single and multisystem forms of LCH have significantly different prognoses, and evaluation for systemic involvement should be undertaken for all patients with cutaneous lesions.
Mutations in BRAF V600E are found in many cases of LCH. A portion of BRAF V600E mutation-negative patients harbor mutations in MAP 2K1.
Introduction
Manifestations of LCH range from relatively benign collections of histiocytes in the skin or bone to multi-organ disease with a higher morbidity and mortality rate. The WHO groups LCH within the larger category of histiocytic/dendritic cell neoplasms (see Tables 4.5 and 4.6) [35, 36].
Table 4.5
WHO classification of hematopoietic/lymphoid tumor subsets, adapted from Campo et al. 2008 [35]
(1) Mature B cell neoplasms |
(2) Mature T cell and NK cell neoplasms |
(3) Hodgkin lymphoma |
(4) Posttransplant lymphoproliferative disorders |
(5) Histiocytic and dendritic cell neoplasms |
– Histiocytic sarcoma |
– Langerhans cell histiocytosis |
– Langerhans cell sarcoma |
– Interdigitating dendritic cell sarcoma |
– Follicular dendritic cell sarcoma |
– Fibroblastic reticular cell tumor |
– Intermediate dendritic cell tumor |
– Disseminated juvenile xanthogranuloma |
Historic classification model | Modern classification |
|---|---|
Eosinophilic granuloma (localized LCH, often of bone) Hashimoto-Pritzker (congenital self-healing LCH) | Single-system LCH (SS-LCH) |
Hand-Schuller-Christian (osteolytic lesions of the skull, diabetes insipidus, and exophthalmos) Abt-Letterer-Siwe (acute diffuse histiocytosis) | Multisystem LCH (MS-LCH) -With “risk organ” involvement (liver, spleen, hematolymphatic, or respiratory system) -Without “risk organ” involvement |
Recently, authors have advocated for a new classification scheme of LCH from the traditional eponyms into single-system (SS-LCH) and multisystem LCH (MS-LCH) (see Table 4.6). Focal osteolytic lesions, previously known as eosinophilic granuloma, and congenital self-healing reticulohistiocytosis (Hashimoto-Pritzker) are now considered variants of SS-LCH.
Epidemiology
Across subtypes, LCH occurs at a rate of about 1–4 per million children [38–40), with rates as high as 9 per million in some studies [41]. Rates are generally higher in children under age 1 (around 5–9 per million infants) [38, 39], as well as in Caucasian and Hispanic populations [38]. Most cases of LCH are sporadic, but familial and twin cases have been reported [42]. Most case series show a modest male predominance.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


