Syndrome
Gene mutation and inheritance
Clinical features
RMS type and/or features
Other associated malignancies
Li–Fraumeni
P53 AD
Early onset malignancy, familial
Anaplastic RMS presents in children <3 years old
• Breast cancer
• Non-RMS tumors
• Brain tumors
• Leukemia
• Colon cancer
• Others
Beckwith–Wiedemann
(1) Mutation/deletion of imprinted genes chromosome 11p15.5
(2) Heterozygous mutation in CDKN21 AD
Pediatric overgrowth syndrome with variable features: macroglossia, omphalocele, hypotonia, neonatal hypoglycemia, prominent nevus simplex
Embryonal RMS
• Wilms’ tumor
• Neuroblastoma
• Hepatoblastoma
• Adrenal carcinoma
NF type I
NF1 AD
Café au lait macules, axillary/inguinal freckling, lisch nodules, sphenoid wing dysplasia, pseudarthrosis, neurofibromas, macrocephaly, developmental delay
Embryonal RMS young age of presentation, genitourinary site
• Optic glioma
• Malignant peripheral nerve sheath tumors
• Meningioma
• Pheochromocytoma
• Pilocytic astrocytoma
Noon an
PTPN11 AD
Short stature, webbed neck, lymphatic malformations, low set ears, VSD, pulmonic stenosis
Embryonal RMS
• Malignant schwannoma
Cos tello
HRAS AD and sporadic
Short stature, macrocephaly, coarse facies, webbed short neck, hypertelorism, thick lips, sparse curly hair, papillomas, loose skin, acanthosis hypertrophic cardiomyopathy, pulmonic stenosis, mitral valve prolapse
Embryonal RMS, most common malignancy in Costello syndrome patients
• Neuroblastoma
• Bladder carcinoma
• Vestibular schwannoma
Prognosis
Prognosis for patients with RMS has improved significantly over the past decade due to multimodal chemotherapy regimens. Relapse-free survival rates are reported in the 70–80% range [7]. Children aged 1–9 have the best prognosis, with infants less than age 1 and older children showing significantly worse outcomes. The Intergroup Rhabdomyosarcoma Study Group (IRSG) recently reported that 5-year failure-free survival (FFS) was 57% for patients less than 1 year, 81% for patients aged 1–9 years, and 68% for patients older than 10 years. Five-year survival for these groups was 76%, 87%, and 76%, respectively [7].
Treatment
Treatment for RMS is multimodal and includes surgical management, chemotherapy, and often radiation. However, these chemotherapeutic regimens are associated with significant toxicity. Advances in the molecular classification of RMS may be paving the way for more targeted therapies and will be reviewed with each histopathologic subtype below [8].
Embryonal RMS (Fusion Negative)
Embryonal RMS is the most common histopathologic subtype, seen most frequently in children ages 0–4 years. It is more common in males (1.5:1) [9]. Embryonal RMS tends to occur on the head and neck or genitourinary (GU) tract. Characteristic genetic changes in embryonal RMS include loss of heterozygosity of 11p15 and gains in chromosome 8. Mutations in NRAS, KRAS, HRAS, and NF1 are present in up to a third of tumors, while mutations in other recognized genes (FGFR4, PIK3CA, CTNNB1, FBXW7, and BCOR) comprise less than 10% of cases [8].
Alveolar RMS (Fusion Positive)
The alveolar subtype of RMS tends to occur in older children/adolescents and comprises about 20% of all RMS cases [10]. It is most common on the trunk and extremities and perianal regions [10]. To be designated as alveolar, over 50% of the tumor must have the alveolar histopathologic features. The distinction between embryonal and RMS can also be made based on genetic classification. The key genetic footprint of classic RMS is the fusion protein between PAX 3 or PAX 7 and the FOXO1 gene [8].
Undifferentiated RMS (Pleomorphic, Anaplastic)
Pleomorphic and anaplastic RMS account for only 2% of all pediatric RMS, seen more commonly in the adult population. One important exception is the anaplastic RMS subtype (anRMS) which has recently been identified to occur frequently in children with Li–Fraumeni syndrome [11]. A diagnosis of anRMS in children under age 36 months should prompt investigation into underlying p53 mutations so that surveillance for other associated secondary malignancies can be undertaken [11].
Genetic Classification and Advances
Recently, extensive genomic analysis of numerous RMS tumors supports that the molecular classification is the most helpful way to categorize these tumors [8]. In their landmark paper, Shern and colleagues identified that the presence or absence of the PAX3/7–FOXO1 gene fusion is the most important prognostic indicator for any RMS. Typically, there is emerging support for molecular classification over histopathologic classification as a more precise predictor of clinical behavior. Fusion positive tumors behave clinically like alveolar RMS, and fusion negative tumors behave as traditional embryonal RMS [12]. Interestingly, the main genetic alterations in all types of RMS seem to be within a common receptor tyrosine kinase/RAS/PIK3CA signaling pathway , either via rearrangement of the PAX gene or by the occurrence of downstream mutations [8]. Advances in this area will likely give rise to more targeted, molecularly based chemotherapies.
Primary Cutaneous RMS
Cutaneous presentations of RMS without another distinct primary site have been rarely documented in case reports with 75% occurring in the pediatric population [13]. Lesions present as grouped nodules usually less than 5 cm in size and may mimic keloid scars, cysts, dermatofibromas, sarcoidosis, basal cell carcinoma, and hematomas [14]. In children, the embryonic and alveolar subtypes predominate with a “small blue cell” pattern by histopathology.
Non-rhabdomyosarcoma
Key Points
Non-rhabdomyosarcoma soft tissue sarcomas [NRMS] may mimic vascular tumors, morphea, or benign fibrohistiocytic tumors on clinical and radiologic exams. A biopsy is required for diagnosis.
Surgical resectability is a key factor in overall prognosis.
Most NRMS occurring early in life have a favorable prognosis if recognized and treated promptly.
The non-rhabdomyosarcoma soft tissue sarcomas represent 3–4% of all pediatric cancers [15, 16]. Classification is based on the International Classification of Childhood Cancers. Prognostic factors for all subtypes include extent of disease (local vs. metastatic), degree of tumor resection, maximal tumor diameter (<5 vs. >5 cm), and tumor grade [1, 17–19].
In all cases, a tissue biopsy is required to ascertain a definitive diagnosis. Tumors may be located deeper in the tissue than clinical exam suggests, so preoperative imaging is suggested. Close collaboration with an oncologist is necessary to facilitate proper cancer staging and treatment. As the lungs are the most frequent site of metastasis, chest imaging is an important component of the evaluation. Additional imaging studies should be dictated by the tumor subtype and exam findings [1].
Once the diagnosis and staging are confirmed, tumor resection is a key component of treatment. The role of neoadjuvant and adjuvant chemotherapy and radiation varies based on patient age, tumor characteristics, and the extent of resection [1, 20–22] and therefore requires the input of an oncologist who is well versed in the treatment of pediatric soft tissue tumors.
Fibrous (Connective Tissue Tumors)
Key Points
Fibrosarcomas present as firm, non-mobile soft tissue masses. They have a bimodal age distribution. Presentation prior to age 2 years is associated with a favorable prognosis.
Dermatofibrosarcoma protuberans is a fibrous tumor which is very rare in children. Consider underlying severe combined immunodeficiency (SCID) syndrome especially in children with multicentric lesions.
Fibrosarcoma
Epidemiology
Fibrosarcoma (FS) is the most common soft tissue tumor occurring in infancy, representing 10% of the pediatric non-rhabdomyosarcoma soft tissue sarcomas [23]. The age distribution is bimodal with the initial peak occurring in infancy and the second in early adolescence. Infantile fibrosarcomas may be congenital or arise in the first few years of life. Most experts suggest that tumors occurring prior to the age of 2 years are of the infantile subtype [24]. The second peak in incidence occurs in the early teens, between 10 and 15 years of age, with tumor characteristics and prognosis similar to those of adult FS [23].
Clinical Characteristics
FS present as rapidly growing superficial or deep soft tissue masses. They have a predilection for the distal extremities in younger children and more axial locations in older patients [23] (Fig. 6.1). Children rarely report associated pain. The overlying skin appears glossy, tense, and erythematous and may ulcerate [23]. Presentations mimicking vascular anomalies including infantile hemangiomas and lymphatic malformations have been reported [25–27]. Unlike infantile hemangiomas which are soft and mobile, FS are infiltrative, fixed to the underlying tissue [26]. Associated hematologic abnormalities include low-level thrombocytopenia and coagulopathy mimicking Kasabach–Merritt phenomenon [26–29].
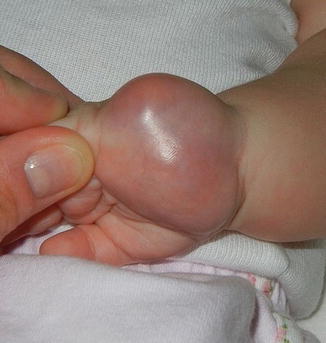
Fig. 6.1
Firm, pink tumor on the palm of an infant: infantile fibrosarcoma (Image courtesy of Jennifer T. Huang, MD)
Laboratory and Imaging Characteristics
Tumor biopsy is required for diagnosis. The cells of FS are spindled and densely packed with frequent mitoses. Highly vascular areas are common. Immunohistochemical staining is variable but is usually positive for vimentin and negative or focally positive for smooth muscle actin, desmin, S-100 protein, or CD34 [26, 30]. When infantile hemangioma is considered in the differential diagnosis, staining for GLUT-1, positive in IH but negative in FS, may be helpful. Infantile and adult FS are histologically identical [24] so molecular diagnostics may further differentiate these entities. Aberrant copies of chromosomes 8, 11, 17, and 20 are all more common in infantile FS [26, 31–33]. Recently the gene translocation t(12;15)(p13;q25) resulting in the fusion protein ETV6-NTRK3, and expression of NTRK3 (TRKC), a tyrosine kinase receptor, has been reported as a more specific marker for infantile FS [26, 30, 32–36].
Prognosis and Treatment
Patient age is the most important prognostic feature. Infantile FS have a >80–90% survival rate [23, 36], with survival falling to 50–60% in older children [23, 37]. In a series of children under the age of 2 years, the 5-year survival rate was 89%, even though only 21% underwent complete surgical resection [36]. For this reason, aggressive or deforming surgeries are only recommended when other treatment modalities have failed [36, 38].
FS are chemoresponsive [36]. Neoadjuvant chemotherapy may allow for a smaller excision with less resultant functional or cosmetic compromise and may be curative [38]. As recurrences of infantile FS tend to be local as opposed to metastatic, some authors recommend careful watchful waiting after chemotherapy as opposed to a surgical intervention.
While there have been reports of spontaneous resolution of infantile FS without treatment [39, 40], this is uncommon and should be reserved for cases of infants <3 months old with poorly operable tumors [36].
LOXO-101 , an experimental drug targeting a tropomyosin-related kinase inhibitor , is a promising new treatment for infantile FS that express the NTRK3 receptor . While only one pediatric case report has been published, the response was dramatic in an otherwise refractory tumor [41]. Additional data is needed to determine the full potential of this medication.
Non-infantile fibrosarcomas require more aggressive therapy given their worse prognosis. Their predilection for axial sites makes complete resection more difficult, increasing the risk of metastasis [23]. In many cases, chemotherapy and radiation are indicated, especially in the setting of incomplete surgical resection [1, 23].
Dermatofibrosarcoma Protuberans
Epidemiology
Dermatofibrosarcoma Protuberans (DFSP) is an uncommon mesenchymal tumor of intermediate malignancy. Patients most commonly present in middle age; less than 200 cases of pediatric DFSP were reported in the literature [42]. A review of pediatric cases revealed no gender predilection. In this group, the mean age at diagnosis was 9 years, and the mean time to diagnosis was 4 years [42].
Clinical Presentation
In both adults and children, DFSP follow an indolent course of slow growth with local invasion and frequent recurrence [42]. Most tumors arise on the trunk, with nearly all congenital cases arising at this site [43]. The proximal lower extremity is also a common location. Tumors of the head and neck or upper extremities are less common.
DFSP may begin as pink macules and may appear vascular (Fig. 6.2). They slowly progress into indurated pale red-blue plaques and then nodules. Additional nodules may develop at the periphery giving a multilobulated appearance [42, 44, 45]. A plaque-like morphology has been described more often in pediatric cases [45]. Plaques are fixed to the skin but are mobile over underlying tissues [43, 45–47]. Tumors typically range between 1 and 5 cm in diameter but may grow significantly larger [44, 46]. They are primarily asymptomatic, but ulceration and pain can occur [42, 46, 47].
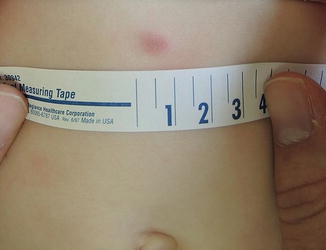
Fig. 6.2
Firm violaceous nodule in a 10-month-old: DFSP (Image courtesy of Ingrid Polcari, MD)
Cases of DFSP arising in children with severe combined immunodeficiency (SCID) syndrome , Fanconi anemia , Shwachman–Diamond syndrome , and Cowden syndrome have been described [48–51]. Of these potential syndromic associations, adenosine deaminase-deficient severe combined immunodeficiency (ADA-SCID) syndrome is the most convincingly linked. In a series of 12 ADA-SCID patients who received skin checks, 8 were found to have DFSP, and 7 had multicentric lesions [48]. Two additional case reports have been published [52, 53]. While the cause of the association is uncertain, authors speculate that increased levels of adenosine and deoxyadenosine produce profibrotic effects and DNA strand breaks in the skin, creating a favorable environment for the development of DFSP [48]. Single cases of DFSP arising in children with Shwachman–Diamond, Fanconi anemia, and Cowden syndrome have been reported, but the associations are not well established.
Clinical mimickers of DFSP include benign lesions such as dermatofibromas, vascular malformations, pilomatricoma, morphea, atrophoderma, anetoderma, scar, keloid, infantile myofibroma, and neurofibroma [42, 54, 55]. The combination of benign appearance, lack of symptoms, and slow growth likely contributes to the often delayed diagnosis.
Laboratory and Imaging Tests
The histopathology of DFSP is characterized by a storiform collection of spindled cells extending through the dermis and into the fat. Immunohistochemical staining is positive for CD-34 and negative for factor XIIIa. A reciprocal translocation T(17;22)(q 22, q 13) is the most common gene rearrangement in pediatric DFSP and may be detected by cytogenetic testing [56].
Prognosis and Treatment
Primary treatment consists of surgical excision with wide margins. Margins of 2–3 cm in children >5 years and of 1–2 cm in younger children are suggested [57, 58]. Multiple procedures are often needed to obtain microscopically clear margins [59].
Mohs excision has been reported as a successful treatment with low risk of recurrence, but anesthetic exposure may limit its use in small children [60–62]. The recurrence rate in adults is reported at 32–76% with most recurrences occurring in the first 3 years [63]. Preoperative imaging, particularly in larger tumors, may help to delineate the tumor dimensions improving successful surgical clearance [64]. Sites of metastasis include the lungs and the lymph nodes, but metastatic disease is rare in children [45, 58, 59].
Imatinib mesylate is a tyrosine kinase inhibitor which has been used to successfully treat DFSP in adults. In children with unresectible or recurrent tumors, it has been reported as a neoadjuvant therapy, decreasing tumor size to allow for complete resection [57].
Borderline or Malignant Vascular Tumors
Key Points
Immune suppression is an essential trigger for all Kaposi sarcoma subtypes.
HIV-associated (epidemic) Kaposi sarcoma has now become the most common KS variant in children.
Kaposiform hemangioendothelioma is a rare vascular tumor of infancy. It is an important cause of Kasabach–Merritt phenomenon which may be fatal even when promptly identified and treated.
Kaposi Sarcoma
Epidemiology
Kaposi sarcoma (KS) is a malignancy of endothelial cell origin occurring in the setting of immunosuppression. It was originally described in elderly males of Eastern European and Mediterranean ancestry (classic KS) but was later identified in an endemic form in portions of Africa and in an epidemic form in patients infected with HIV/AIDS. Iatrogenic cases are attributed to immune suppression following solid organ transplantation.
Pediatric KS is rare in most of the world but comprises 25–50% of pediatric soft tissue sarcomas and 2–10% of all pediatric cancers in portions of Eastern and Southern Africa [65–68]. All subtypes of KS have been reported in children [69–71]. Prior to the late 1980s, endemic KS was the most common variant seen in children, but now HIV-associated (epidemic) KS predominates [68]. Pediatric cases of classic KS remain rare with less than 50 reported cases, all occurring within the Mediterranean basin [68, 72].
Pediatric KS has a predilection for males. The mean age of onset is reported at 6.6 and 8.8 years for endemic and epidemic variants, respectively, but early onset is not uncommon [73, 74]. The timing of onset of iatrogenic KS is dependent upon the age at transplantation. Classic KS has been described in children ranging from infancy to the teen years [68].
Clinical Characteristics
The clinical presentation of pediatric KS varies by subtype. Skin and mucosal lesions, when present, begin as purple or brown-red patches that grow into plaques and nodules. Lesions are usually painless but may Koebnerize in areas of past trauma or infection [74]. Classic KS favors acral sites in children as it does in adults. Children with endemic KS are more likely to present with lymphadenopathy and less commonly with skin or mucosal involvement, but data is limited [73]. Lymphoma, bacillary angiomatosis, and other disseminated infections associated with lymphadenopathy are clinical mimics [75].
In a series of children with primarily epidemic KS, three patterns of disease were described (listed in order of frequency):
- 1.
KS of the head and neck with localized or generalized lymphadenopathy, 39% with skin lesions
- 2.
KS of the genital area with inguinal lymphadenopathy, 57% with skin lesions
- 3.
Solitary tumors localized to the extremities or the abdominal organs [74]
Laboratory and Imaging
The histopathology of KS is characterized by collections of spindled cells with slit-like vascular spaces and erythrocyte extravasation. Cells stain positive for the vascular endothelial markers CD31, CD 34, and factor VIII. The most definitive marker is positive staining for human herpesvirus 8 latency-associated nuclear antigen, present in all cases of KS [76].
HHV8 is a necessary factor in the pathogenesis of KS [66, 77]. Most individuals infected with HHV8 do not develop KS, suggesting that an acquired or inherited immunodeficiency which increases susceptibility to the virus is a necessary cofactor [68, 78].
Radiographic imaging is useful in determining disease extent. Based on sites of suspected involvement, contrast-enhanced CT or MRI is suggested. In both modalities, tumors show strong post-contrast enhancement. Scintigraphy (lymphoscintigraphy) with sequential thallium and gallium scanning may be employed to differentiate KS from infectious mimics [75].
Treatment and Prognosis
There are no consensus guidelines on the treatment of pediatric KS. In classic, epidemic, and endemic forms, chemotherapy is indicated for systemic involvement [68, 72, 79]. Intralesional chemotherapy may be considered for localized disease [68]. In children with epidemic KS, the combination of HAART with chemotherapy appears to be the most beneficial [80]. Notably, children with epidemic KS who are treated with HAART have an elevated risk of developing immune reconstitution inflammatory syndrome (IRIS) [68] and must be monitored for this complication.
Children with iatrogenic KS have a better overall prognosis than those with epidemic KS [73]. Some cases of iatrogenic KS have resolved with transition to sirolimus-based immunosuppression. Sirolimus , an mTOR inhibitor, may exert its effect by inhibiting angiogenesis and autocrine growth factor signaling within the tumor [79]. Based on a mouse model, mTOR inhibitors may be effective in all KS subtypes and better tolerated than other systemic agents, but further human trials are needed to make accurate conclusions [79, 81].
Survival in KS is dependent on HIV/AIDS status and stage of disease at presentation. Access to care may be limited and likely contributes to worse outcomes in resource-poor areas.
Kaposiform Hemangioendothelioma
Epidemiology
Kaposiform Hemangioendothelioma (KHE) is a rare, borderline malignant, infiltrative vascular tumor. KHE can cause life-threatening complications secondary to associated Kasabach–Merritt phenomenon (KMP). The estimated prevalence of KHE is 0.91 per 100,000 children with a slight male predominance ~1.33:1 [82]. Over 90% of cases occur in infancy, usually in the first 3 months of life.
Clinical Presentation
KHE typically presents as a solitary lesion, though multifocal presentations have been described. KHE is infiltrative and may cross tissue planes from the dermis to the underlying subcutaneous tissue, muscle, and bone. The most common locations include, in order of frequency, the extremities, torso, and the head and neck. Extension to the retroperineum is often observed [82, 83]. Cutaneous features include a solitary, indurated, violaceous, or erythematous to purpuric plaque or tumor. Hypertrichosis and hyperhidrosis may also be appreciated (Fig. 6.3a).
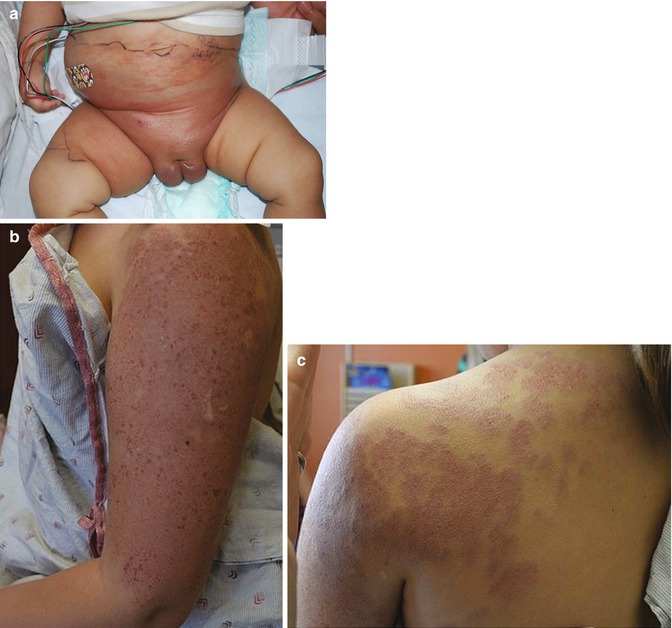
Fig. 6.3
(a) KHE in the perineum and lower abdomen (Image courtesy of Kristen Hook, MD). (b and c) Erythematous, indurated plaque of the upper extremity and shoulder of a 9-year-old girl: large tufted angioma previously treated with oral sirolimus
KMP is a consumptive coagulopathy which is specifically observed in the setting of KHE or tufted angioma and is usually present at the time of diagnosis. Tufted angioma (Fig. 6.3b) shares similar clinical and histopathologic features with KHE, and the two entities are considered to represent a spectrum of severity. KMP is a life-threatening condition due to sequestration of platelets within the tumor resulting in thrombocytopenia. Though the exact etiology is not well understood, it is thought that turbulent blood flow within the slit-like spaces of the KHE may lead to consumption of platelets and, when severe, can result in profound thrombocytopenia, elevated D-dimer levels, and low fibrinogen.
Initial recommended workup includes a complete blood count, coagulation studies, and baseline MRI imaging (with and without contrast) along with tissue biopsy to confirm the diagnosis.
Prognosis
There is a lack of outcome data related to the management of KHE and KMP. Historically, mortality rates in the 1980s were reported in the range of 30%, usually due to hemorrhage or other complications related to KMP [84]. However with earlier diagnosis and improved medical management, outcomes seem to have improved significantly.
Treatment
An interdisciplinary consensus statement on the management of KHE was proposed in 2013 [85]. At that time, authors advocated for initial treatment with vincristine and systemic steroids. More recently, the use of oral sirolimus in the management of KHE and KMP has been very promising [86] and may be preferable as an initial treatment due to the more favorable side effect profile and ease of administration. Sirolimus has also been reportedly successful in the late stage of KHE with softening of the tumor and improvement in pain and joint contracture [87]. A study comparing sirolimus and vincristine in the management of KHE is currently underway [86].
Neural
Key Points
Malignant peripheral nerve sheath tumors usually arise after puberty and are strongly associated with neurofibromatosis 1.
MPNST may be difficult to discern clinically and radiographically from a growing plexiform neurofibroma.
Malignant Peripheral Nerve Sheath Tumors (MPNSTs) (Synonyms Neurofibrosarcoma, Neurogenic Sarcoma, Malignant Schwannoma)
Epidemiology
MPNSTs form from the nerve sheaths of larger nerve trunks [21, 88, 89]. They represent 5–10% of all soft tissue tumors [90]. Less than 20%, however, present in children and usually arise after puberty [21, 91].
Neurofibromatosis type 1 is a closely linked risk factor. Even in the absence of NF-1, most MPNSTs demonstrate a pathogenic truncating mutation in the NF-1 gene [92, 93]. Approximately 10% of NF-1 patients will develop a MPNST, usually within a plexiform neurofibroma (NF), but incidence estimates vary widely [94, 95] The mean age of MPNST onset in NF-1 positive patients is 27 years, compared with 40 years in patients who are NF-1 negative [96].
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


