Fig. 9.1
Pathophysiology of acute GVHD. IL 1 interleukin 1, IFN γ interferon γ, LPS lipopolysaccharide, Treg regulatory T cell, Th1 T-helper cell, CTL cytotoxic T lymphocyte [2]. Reprinted from The Lancet, Vol. 373, number 9674, Ferrara JLM, Levine JE, Reddy P, Holler E, Graft-versus-host disease, pages 1550–1561, © 2009, with permission from Elsevier
While the pathophysiology of chronic GVHD is not fully understood, the diversity of clinical phenotypes and the discovery of autoantibodies and genetic polymorphisms similar to patients with classic autoimmune disorders suggest a more complex immune reaction than that seen in acute GVHD [4]. Chronic GVHD typically follows acute GVHD, yet it may also occur de novo. In the Task Force Report from the National Institutes of Health Consensus Development Project on Criteria for Clinical Trials in Chronic Graft-Versus-Host Disease, Cooke et al. propose another three-phase model [5]. Similar to acute GVHD, the first phase is characterized by tissue injury, inflammation, and activation of donor T cells. The second phase involves diffuse, nonspecific inflammation as donor T cells migrate through inflamed, leaky vasculature and lymphatics. Activation of the adaptive immune system also takes place; however, maturation of T cells in a dysfunctional thymus—one that is aging or has been damaged by conditioning or acute GVHD rendering it incapable of negative selection—leads to loss of central tolerance. Peripheral tolerance is also diminished due to an imbalance between regulatory T cells (Tregs) and alloreactive T cells. In the final phase, aberrant tissue repair and fibrosis may occur, leading to irreversible organ damage [5].
Overall, risk profiles for acute and chronic GVHD are similar [6]. GVHD occurs most commonly after allogeneic HSCT, due to inherent HLA disparity between the recipient and donor. Though less common, GVHD may also arise following autologous HSCT (the recipient and donor are the same patient) [7] and also after solid-organ transplant (most commonly in small bowel or liver transplantation) [8–11]. GVHD following autologous HSCT is hypothesized to result from sensitization of the harvested cells during processing and storage prior to reintroduction to the patient. In solid-organ transplant, co-transplantation of alloreactive immune cells residing in the donor tissues initiates the GVHD cascade [9]. In allogeneic HSCT and solid-organ transplants, the most significant risk factor is HLA mismatch between donor and recipient. Risk for GVHD may be higher with HLA mismatch at the HLA-A or -B locus. It is also important to note that acute GVHD increases the risk of chronic GVHD by 11-fold [12]. Additional clinical, genetic, and biomarker-based risk factors are listed in Table 9.1 [13–22].
Table 9.1
Risk factors for acute and chronic GVHD
Host variables |
• Malignancy as indication for transplant, as well as features of more advanced disease (WBC >50 × 109/L and cytogenetic abnormalities t(4;11), t(9;22), and hypodiploidy) [13–16, 22] • Prior damage to gut (viral illness, prolonged fasting, chemotherapy) [15] |
Donor or graft variables |
• HLA mismatch [14] • Unrelated donor [14] • ABO blood group mismatch [14] • Graft source: allogenic HSCT (PBSC > BM > UCB) > autologous HSCT > solid organ transplant [19, 21, 22] • Graft with high CD34+ cell dose or low regulatory T-cell content [15] |
Other variables |
• Genetic polymorphisms within genes encoding for innate immunity, or inflammatory/immunoregulatory proteins in either donor or host [15] • Prior acute GVHD (increases the risk for chronic GVHD) [20] |
Incidence of GVHD
Key Points
The lowest rates of acute and chronic GVHD are among recipients of HSCT from related donors and from umbilical cord donors.
The incidence of acute and chronic GVHD in children is approximately half that of adults.
In the United States, over 1500 allogeneic HSCTs are reported annually in patients less than 20 years old [23]. The incidence of GVHD within this population ranges widely depending on risk factors, most notably HLA compatibility and stem cell graft source (e.g., bone marrow, peripheral blood, or umbilical cord blood). About two-thirds of allogeneic HSCT in children are from unrelated donors and the majority of children receive HSCT derived from the bone marrow or cord blood [23].
In those receiving unrelated donor HSCT, Grade II–IV acute GVHD has been reported in 40% of cord blood and 85% of bone marrow HSCT [12, 16, 21, 24]. There is greater immune tolerance, and thus lower incidence of acute GVHD, associated with cord blood HSCT compared to bone marrow HSCT for similar levels of HLA mismatch. In those receiving related donor HSCT, the incidence of grade II–IV acute GVHD is significantly lower, reported in about 25% of those receiving grafts from HLA-identical siblings, with equivalent rates between bone marrow and peripheral blood cell sources [21].
The incidence of chronic GVHD varies by these same factors. Chronic GVHD has been reported in as few as 6% of recipients of sibling-related umbilical cord blood HSCT and up to 65% of recipients of mismatched peripheral blood stem cell HSCT [19, 25–27]. The onset of chronic GVHD in relation to acute GVHD is progressive in 30–40%, quiescent (e.g., acute GVHD occurred but resolved prior to chronic GVHD onset) in 30–40%, and de novo in 20–30% [28].
In general, the incidence of GVHD in children is about one-half that of adult populations [20, 29, 30]. Decreased incidence may be attributed to more frequent use of cord blood as a stem cell source in children, nonmalignant indications for HSCT, limited history of prior infections, and overall improved state of health in children [14].
Clinical Features of GVHD
Key Points
Acute GVHD most often occurs within months after HSCT, but may occur at any point and can overlap with chronic GVHD.
Acute GVHD primarily involves the skin, liver, and gastrointestinal tract.
Chronic GVHD may have more diffuse, often irreversible, organ involvement.
Although acute GVHD most often occurs within 1–2 months after HSCT [2, 16], the diagnosis can be made at any point after HSCT. Because time-based criteria are currently less emphasized, there is a greater emphasis on clinical features in making the diagnosis of acute GVHD [2, 6, 13].
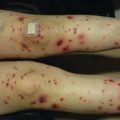
Acute GVHD most commonly targets the skin, liver, and gastrointestinal tract [4, 31]. The skin is the most frequently affected organ and is often the first involved [2]. The classic rash is pruritic, may be painful, and is characterized by erythematous macules and papules coalescing on the trunk and extremities (often sparing the scalp), resembling the morbilliform rash of measles (Fig. 9.2). Acral involvement is common (Fig. 9.3). In severe GVHD, bullae and desquamation may develop, and with extensive involvement may resemble toxic epidermal necrolysis (Fig. 9.4). Gastrointestinal symptoms include nausea, vomiting, anorexia, abdominal pain, and diarrhea [2].
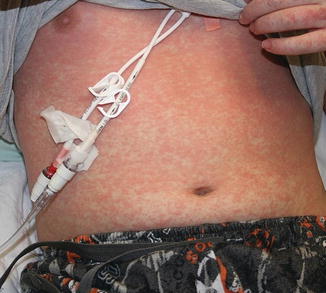
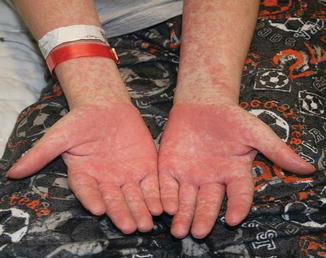
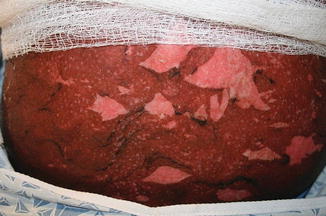
Fig. 9.2
Acute GVHD presenting as a morbilliform skin eruption
Fig. 9.3
Acral involvement in acute GVHD
Fig. 9.4
Toxic epidermal necrolysis -like acute GVHD
Children tend to develop symptoms of chronic GVHD at a median of 6 months after HSCT [30]. About 40% of these patients manifest extensive disease; the remainder experience limited involvement of the skin, liver, or both [19]. Again, the skin is the most commonly affected organ, with cutaneous features in 65–80% of children with chronic GVHD, followed by oral lesions in half, liver disease in a third, and gastrointestinal involvement in 25–60% [19, 32]. Involvement of the lungs, eyes, and/or musculoskeletal system is not uncommon [6, 19, 33].
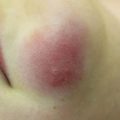
Cutaneous chronic GVHD (particularly sclerotic forms) is often preceded by peripheral edema in children (Fig. 9.5) [34]. Chronic GVHD has a propensity to affect the mouth, nails, and areas of friction such as the waistband, while the face and digits are rarely involved. Affected limbs can become bound-down and restricted in width. Dyspigmentation is almost universal and vitiligo is a known, but less common, presentation (Fig. 9.6) [30, 34]. The depth of sclerotic disease ranges from superficial lichen sclerosus-like lesions to dermal fibrosis and myofascial involvement (Figs. 9.7 and 9.8) [34]. Of note, fasciitis and myositis can arise independent of skin involvement and predispose to permanent contractures [6]. Eczematous and ichthyosiform features can be found in sclerotic and nonsclerotic disease and may be more common in children than adults (Fig. 9.9) [34]. The reported incidence of lichenoid lesions varies widely; they may be more common in steroid-refractory chronic GVHD [33–35]. It is important to recognize that multiple morphologies may be present in an individual patient, and thus a thorough skin examination is imperative.
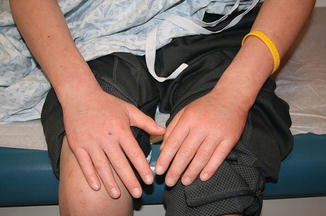
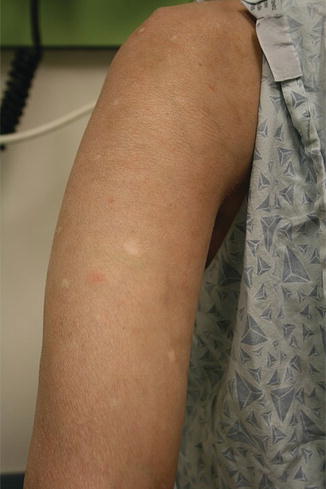
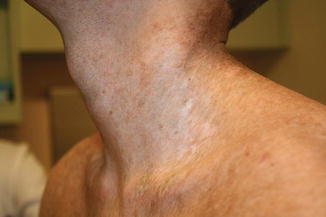
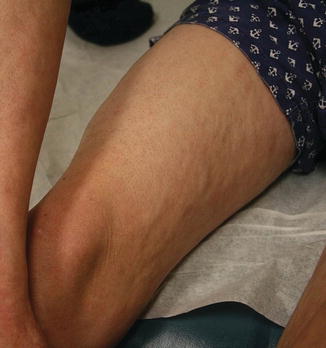
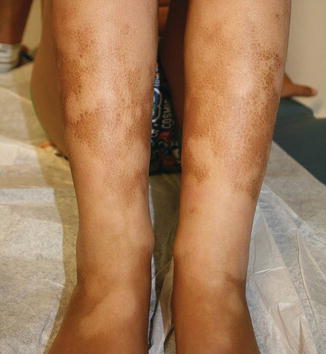
Fig. 9.5
Acral edema as an early sign of sclerotic chronic GVHD
Fig. 9.6
Dyspigmentation (both hyperpigmentation and hypopigmentation) in a child with chronic GVHD
Fig. 9.7
Morpheaform sclerotic chronic GVHD
Fig. 9.8
Sclerotic chronic GVHD with myofascial involvement
Fig. 9.9
Eczematous or ichthyosiform presentation of chronic GVHD
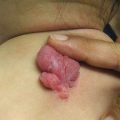
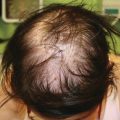
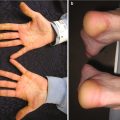
Mucosal, hair, and nail findings may also be appreciated during dermatologic examination. The eyes may be less commonly affected by chronic GVHD in children than other organs, though one study reports involvement in 50% of patients [19, 33]. In these patients, lacrimal dysfunction leads to conjunctivitis [5, 6]. Oral lesions may be erythematous, reticular, or ulcerative; they are infrequently painful, leading to underreporting [32]. Involvement of the oral mucosa often corresponds to genital findings including lichen planus-like features, lichen sclerosus-like features, clitoral and vaginal scarring in females, and phimosis and urethral stricture in males [6]. Focal or diffuse alopecia may occur in up to 50% of children and can be scarring or nonscarring (Fig. 9.10) [30]. Nails are affected in up to 45% of children, with periungual erythema and/or dystrophy [30, 34]. Pterygium inversum unguis , in which the distal nail bed adheres to the nail plate, is common in severe chronic GVHD and is associated with a higher risk of lung involvement in children (Fig. 9.11) [34].
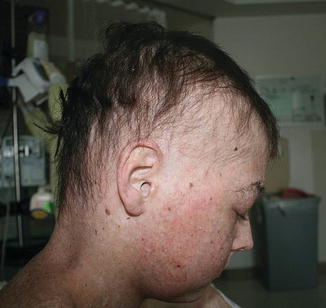
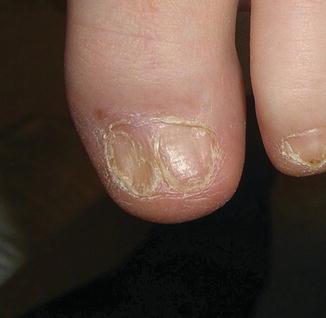
Fig. 9.10
Alopecia is common in children with chronic GVHD
Fig. 9.11
Pterygium nail deformity may be a harbinger of severe lung involvement in children with chronic GVHD
Though outside of the spectrum of dermatologic care, physicians should be aware of other organ systems involved in chronic GVHD. Chronic pulmonary inflammation can lead to bronchiolitis obliterans syndrome [6]. Multifactorial gastrointestinal changes (e.g., scarring, altered motility) can result in decreased intake, poor absorption, and failure to thrive. As in acute GVHD, nonspecific hyperbilirubinemia and transaminitis may also manifest in chronic GVHD [6].
Differential Diagnosis for GVHD
Given the comorbidities of patients at risk for GVHD and the various morphologies of GVHD in its acute and chronic forms, the differential diagnosis for GVHD is broad and includes infectious and inflammatory etiologies.
Bacterial and viral exanthems occur more commonly in children, and solid-organ transplant and HSCT recipients are at increased risk for HHV6 and HHV7 reactivation [36], making infectious etiologies important to consider in patients with acute GVHD. Signs and symptoms of infection typically accompany the classic childhood exanthems and the distribution and evolution of the rash may be helpful in differentiating these from acute GVHD. The viral exanthem of HHV6 is characterized by erythematous macules and papules surrounded by white halos, which begin on the trunk and spread to neck and proximal extremities [36]. It is accompanied by high fever (101–106 °F) and resolves over several days. Atopic dermatitis and allergic contact dermatitis may present with similar pruritic, papular eruptions, though typically without associated systemic signs and symptoms.
Engraftment syndrome is an early complication of HSCT, occurring within 96 h of granulocyte recovery (absolute neutrophil count of ≥500μL for 2 consecutive days) and characterized by fever >38.3 °C without source of infection, rash >25% body surface area (BSA) that is not attributable to medication, weight gain of ≥2.5% of baseline and albumin drop to 90% of pretransplant levels, and non-cardiogenic pulmonary edema [37, 38]. Additional features such as hepatic dysfunction with total bilirubin ≥2 mg/dL or transaminase ≥2 times normal, renal insufficiency with serum creatinine ≥2 times baseline, and transient idiopathic encephalopathy have also been described in adults, though these are less common in children [38]. Given that granulocyte recovery typically takes place 8–27 days following HSCT [39], it can be difficult to distinguish engraftment syndrome from hyperacute GVHD if distinguishing features are not present.
Clinical features of acute GVHD may mimic the range of drug reactions, including morbilliform drug eruptions, drug reaction with eosinophilia and systemic symptoms, radiation-recall dermatitis, toxic erythema of chemotherapy (TEC) , and Stevens-Johnson syndrome / toxic epidermal necrolysis [4, 14]. TEC is a spectrum of cutaneous reactions to chemotherapeutic agents most commonly presenting with erythema and tenderness of the palms, soles, and flexural regions including the axillae and groin [40]. There may be an increased incidence of TEC with conditioning regimens including busulfan and fludarabine, with a median onset of 22 days after dose administration [41]. See Chap. 7 for a more detailed discussion of TEC. Drug hypersensitivity reactions to non-chemotherapeutic agents should also be considered, yet they tend to occur more in adults. Drug reactions typically occur between 1 and 14 days of initiating a drug, manifesting as a morbilliform rash on the trunk, which spreads to the extremities, and less commonly involves the face, palms, or soles. Comparatively, GVHD is more likely to have acral and facial involvement, follicular prominence, and concurrent diarrhea and hyperbilirubinemia. Radiation-recall dermatitis should also be considered in the setting of total-body irradiation or prior sunburn followed by methotrexate for GVHD prophylaxis.
Lichenoid papules of chronic GVHD may appear similar to lichen planus or lichenoid drug eruption. Voriconazole-induced phototoxicity may present as a macular erythematous rash suggestive of chronic GVHD (Fig. 9.12) [42]. Morphea, scleroderma, lichen sclerosus, eosinophilic fasciitis, atrophoderma of Pasini and Pierini, discoid lupus erythematosus, and vitiligo are all within the differential for sclerotic or dyspigmented GVHD lesions. Alopecia areata, telogen effluvium, and anagen effluvium may produce hair loss in patients also at risk for GVHD.
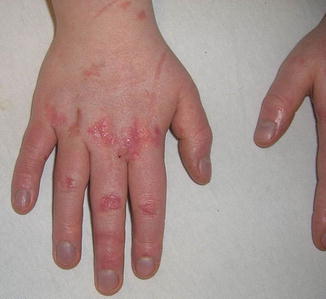
Fig. 9.12
Erythematous , scaly papules in photodistributed locations as a result of voriconazole phototoxicity
Histopathology and Laboratory Evaluation of GVHD
Key Points
Histopathologic features of acute and chronic GVHD are nonspecific.
Biopsy may be helpful for children with distinctive but not clinically diagnostic features of chronic GVHD.
Biopsy is often not necessary to diagnose GHVD. Though skin biopsies may confirm a diagnosis of acute GVHD if clinical suspicion is high, the histologic findings are nonspecific and many of the differential diagnoses show similar features. Histologic findings include sparse lymphocytic interface and perivascular inflammation with variable degrees of adnexal extension. Dyskeratosis, spongiosis, lymphocytic exocytosis, and satellitosis may also be present. In addition to lymphocytic infiltration, eosinophils may be noted, making it difficult to distinguish acute GVHD from drug hypersensitivity reaction. In more severe acute GVHD, subepidermal clefting and full-thickness epidermal necrosis may be seen, mimicking toxic epidermal necrolysis [43, 44]. Thus, biopsy can be unhelpful or misleading if wrongly interpreted and clinical observation with close attention to time course, evolution of disease, and response to withdrawal of a potential offending agent is key in making an accurate diagnosis [45–50]. Histopathology of chronic GVHD is only required for diagnosis of chronic GVHD if features are distinctive but not diagnostic [6]. Features of chronic GVHD vary by clinical manifestation, including epidermal orthohyperkeratosis, hypergranulosis, and acanthosis for lichen-planus-like disease; thickening and homogenization of collagen bundles or pandermal sclerosis with overlying interface changes for morphea-like disease; and homogenization with overlying interface changes for lichen sclerosis-like disease [51].
Additional lab testing is nonspecific for GVHD. Patients with acute GHVD may have hyperbilirubinemia and/or transaminitis [2]. Peripheral eosinophilia has been noted in about half of children with chronic GVHD prior to disease onset [52]. Eosinophilia can be present in patients without chronic GVHD, however, particularly in association with eczema or drug hypersensitivity [35].
Classification of GVHD
Key Points
Staging of acute GVHD relies on the extent of skin, liver, and gut involvement.
The global severity score for chronic GVHD includes the evaluation of eight organ systems.
Proper staging is necessary for prognosis and therapeutic decision making.
Proper classification of acute GVHD is important, as this largely directs therapy. In 1974, Glucksberg devised the original staging system for acute GVHD, which was later modified during the Keystone Conference in 1994 [53]. The Keystone staging attempted to classify acute GVHD based upon the extent of skin, liver, and gut involvement, but the staging of pediatric gut GVHD was not discussed during the Keystone Conference, and stool output varies considerably between children and adults. The current proposal set forth by the University of Michigan and now utilized by the Mount Sinai Acute GVHD International Consortium redefines the Keystone criteria based on volume of diarrhea per kilogram of body weight, rather than absolute volume (Table 9.2) [54]. An additional consideration when staging pediatric acute GVHD is the difference in the distribution of body surface area between adults and children, as children have relatively larger heads and smaller extremities than adults.




Stage | Skin | Liver (bilirubin) | Upper GI | Lower GI (stool output per day) |
|---|---|---|---|---|
0 | No GVHD rash | <2 mg/dL | No or intermittent nausea, vomiting, or anorexia | <10 mL/kg/day or < 4 episodes/day |
1 | Rash <25% BSA | 2–3 mg/dL | Persistent nausea, vomiting, or anorexia | 10–19.9 mL/kg/day or 4–6 episodes/day |
2 | Rash 25–50% BSA | 3.1–6 mg/dL | 20–30 mL/kg/day or 7–10 episodes/day | |
3 | Rash >50% BSA | 6.1–15 mg/dL | >30 mL/kg/day or >10 episodes/day | |
4 | Generalized erythroderma + bullae | >15 mg/dL | Severe abdominal pain ± ileus or grossly bloody stool (regardless of stool volume) | |
Grade a | ||||
0 | None | None | None | None |
I | Stages 1–2 | None | None | None |
II | Stage 3 | Stage 1 | Stage 1 | Stage 1 |
III | Stage 0–3 | Stage 2–3 | Stage 0–1 | Stages 2–3 |
IV | Stage 4 | Stage 4 | Stage 0–1 | Stage 4 |
BSA body surface area, GI gastrointestinal
aGrade is based on most severe target organ, regardless of presence/absence of other organ involvement
Related posts:
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
