Fig. 5.1
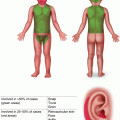
Numerous small skin-colored, dome-shaped papules on the neck, consistent with early basal cell carcinomas in Gorlin syndrome
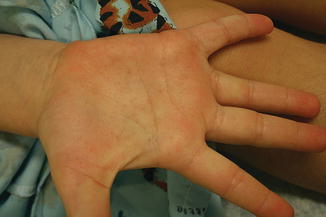
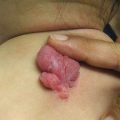
Fig. 5.2
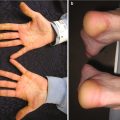
Palmar pits , which can be subtle clinically but represent a major criterion for Gorlin syndrome
Major criteria | Minor criteria |
|---|---|
BCC at age <20 years or multiple BCCs higher in number than would be expected based on exposures | Ocular abnormalities (i.e., hypertelorism, strabismus, congenital cataracts) |
Odontogenic keratocyst during childhood | Skeletal anomalies (i.e., bifid or other rib abnormalities, vertebral fusion, short fourth metacarpals, polydactyly) |
Medulloblastoma | Macrocephaly, frontal bossing |
Calcification of the falx cerebri | Cleft lip/palate |
Palmar or plantar pitting | Fibroma of ovary or heart |
Family history of Gorlin syndrome in a first-degree relative | Abdominal cysts containing lymphatic fluid |
With regard to management for these patients, treatment for each individual BCC is similar to that for any other BCC, typically consisting of electrodessication and curettage (ED&C), curettage alone, excision, or Mohs micrographic surgery for nodular subtype or lesions on sites that are cosmetically sensitive and/or at higher risk for recurrence. When treating multiple BCCs within a localized area, providers should consider field treatment such as photodynamic therapy (PDT) or topical chemotherapy such as 5-fluorouracil cream for superficial lesions. For numerous, locally invasive, recurrent, or metastatic BCCs, one may also consider systemic treatment with the smoothened receptor inhibitors such as vismodegib and sonidegib, although there is currently limited data on safety and efficacy in children [8–10].
These patients require close surveillance with routine full skin examinations every 6–12 months, radiographic surveys to assess for odontogenic keratocysts of the jaw, and counseling regarding importance of strict sun protection. The treatment team should be multidisciplinary and may include dentistry, neurology, orthopedic surgery, ophthalmology, and genetics, in addition to dermatology. While radiographs may be necessary to screen for odontogenic keratocysts or brain tumors, ionizing radiation, e.g., radiologic imaging and radiation therapy, should be used with caution in these patients as it has been shown to significantly increase risk for development of BCCs within the treatment area [11, 12]. With appropriate treatment and no internal malignancy, most patients with nevoid basal cell carcinoma syndrome can expect a normal lifespan.
Bazex Syndrome
Bazex syndrome , also known as Bazex-Dupré-Christol syndrome , is a rare genodermatosis associated with development of multiple BCCs with onset as early as the first or second decade of life [13]. It is thought to have an X-linked dominant inheritance pattern, although the causative gene remains unknown. Other characteristic cutaneous features include hypotrichosis (which is often the presenting sign as it may become apparent early in childhood), hypohidrosis, and follicular atrophoderma of the dorsal aspect of hands and feet [2, 14, 15]. While Bazex syndrome demonstrates significant overlap in clinical features with Rombo syndrome, BCCs in Rombo syndrome tend to develop during adulthood [16]. Management for patients with Bazex syndrome should emphasize education on sun protection as well as regular full skin checks with dermatology.
Squamous Cell Carcinoma
Squamous cell carcinoma (SCC) is the second most common form of NMSC in adults. Many consider keratoacanthomas , which classically present as rapidly enlarging nodules with a central crater containing keratin, to be on a spectrum with SCC. While they may be indistinguishable from SCC histologically, they have a tendency to erupt and then spontaneously regress clinically. Similar to BCC, SCCs are also rare in childhood and should prompt evaluation for predisposing factors. In this section, we will discuss a single genodermatosis that has been associated with development of keratoacanthomas during childhood, Ferguson-Smith syndrome, as well as give brief mention to a second entity that can have similar findings in adults, Muir-Torre syndrome .
Ferguson-Smith
Ferguson-Smith syndrome, also known as multiple self-healing squamous epitheliomas , is characterized by multiple keratoacanthomas , as its name suggests. It has an autosomal dominant inheritance pattern and results from TGFBR1 or ALK5 mutations , resulting in a loss of function mutation in TGFbeta1 [17]. The clinical presentation includes anywhere from a few to hundreds of keratoacanthomas in adolescence or early adulthood that grow rapidly before resolving, often leaving deep scars. The disease is not associated with photosensitivity, but keratoacanthomas have a propensity to develop on sun-exposed areas. Given the spontaneous regression of keratoacanthomas, the lifespan of these patients is typically not affected.
Muir-Torre syndrome is another condition that can be associated with multiple keratoacanthomas, though the tumors typically appear in adulthood. It is an autosomal dominantly inherited condition characterized by sebaceous gland neoplasms and internal malignancies. It is considered a phenotypic variant of hereditary nonpolyposis colorectal cancer (HNPCC) that results most commonly from mutations in mismatch repair genes MLH1 and MSH2 [18]. Sebaceous adenomas, the most common skin neoplasm, are considered benign and usually present in the fifth decade but can present in adolescence [19]. Multiple keratoacanthomas do not usually occur until adulthood. The presence of sebaceous neoplasms in children should prompt screening for Muir-Torre and associated gastrointestinal malignancies [20].
Melanoma
In addition to squamous cell carcinoma in the setting of immunodeficiency, melanoma is a malignancy of the skin with potential to metastasize to internal organs, leading to death. However, when detected early, local surgical excision can be curative. There are a number of factors associated with increased risk for development of melanoma in both children and adults, including, but not limited to: skin type, sun exposure, family history, number and type of nevi, and atypical nevi [2, 15]. In addition, there are several genodermatoses associated with increased risk of melanoma including a familial mole syndrome and xeroderma pigmentosum (to be discussed in a later section), as shown in Table 5.2. In this section, we will focus on familial atypical multiple mole-melanoma syndrome, a condition associated with atypical nevi and increased risk for melanoma.
Table 5.2
Genodermatoses associated with cutaneous malignancy in childhood
BCC | Nevoid basal cell carcinoma syndrome ( Gorlin) Bazex syndrome (Bazex-Dupré-Christol) Xeroderma pigmentosum Oculocutaneous albinism |
SCC | Ferguson-Smith syndrome Xeroderma pigmentosum Kindler syndrome Bloom syndrome Rothmund-Thomson syndrome Oculocutaneous albinism Epidermodysplasia verruciformis Epidermolysis bullosa Incontinentia pigmenti |
Melanoma | FAMMM syndrome (familial atypical multiple mole-melanoma syndrome) Xeroderma pigmentosum |
Familial Atypical Multiple Mole-Melanoma Syndrome
Familial atypical multiple mole-melanoma (FAMMM) syndrome , also known as atypical mole syndrome, is an autosomal dominantly inherited genodermatosis that is most commonly associated with a mutation in CDKN2A, which encodes p16. Typical skin findings include 50 or more atypical nevi (larger in size, darker in color, and/or having irregular borders compared to banal nevi) that develop in childhood or early adulthood, with nevus counts in some individuals reaching several hundred [21]. These patients are at significantly increased risk for development of cutaneous melanoma beginning in their teenage years, as well as ocular melanoma and other internal malignancies, such as pancreatic cancer, with particular CDKN2A mutations [2, 22]. Routine screening skin exams and consideration of total body photography, in addition to sun protection, are integral management strategies to allow for early detection of melanoma in these patients and their families.
Genodermatoses Associated with Photosensitivity and Increased Risk of Skin Cancer
Introduction
Photosensitivity is an important feature of many genodermatoses, some of which are associated with an increased risk for skin cancer and other malignancies. There are multiple underlying mechanisms for the increased risk in skin cancer, which include defects in DNA repair, cell matrix instability, and melanin production, as detailed in Table 5.3.
Table 5.3
Genodermatoses with photosensitivity with underlying defect in DNA repair
Genodermatosis with photosensitivity | DNA repair gene defect |
Xeroderma pigmentosum | Nucleotide excision repair: XPA-XPG, XPV UV-irradiated DNA repair: XPV |
Xeroderma pigmentosum/Cockayne overlapa | Nucleotide excision repair: XPG, sometimes XPB or XPD |
Cockayne syndromea | Nucleotide excision repair: CSA, CSB |
Trichothiodystrophya | Nucleotide excision repair: TTDA, TTDN |
Bloom syndrome | Helicase: RECQL4 |
Rothmund-Thomson syndrome | Helicase: RECQL2 |
Key Points
Xeroderma pigmentosum (XP) is a genodermatosis associated with photosensitivity secondary to defects in DNA mismatch repair and increases the risk for NMSC and melanoma.
Bloom and Rothmund-Thomson are genodermatoses associated with photosensitivity secondary to defects in DNA helicase genes and increase the risk for NMSC.
Kindler syndrome is associated with photosensitivity due to cell matrix instability, and oculocutaneous albinism is associated with photosensitivity due to a defect in melanin biosynthesis. Both increase the risk of NMSC.
DNA Repair
Table 5.3 lists genodermatoses with photosensitivity with underlying defect in DNA repair.
Xeroderma Pigmentosum
Xeroderma pigmentosum (XP) is an autosomal recessively inherited disease characterized by exquisite sensitivity to ultraviolet (UV) radiation. Mutations in eight genes (XPA to XPG and XPV), which correspond to complementation groups that assist in DNA repair, have been identified. The repair rate of damaged DNA depends on the specific mutation, resulting in a range of severity in disease presentations. The incidence of disease ranges from 2.3 per million in Western Europe to 1 per 20,000 in Japan and depends on the demographic of the population [23].
There are two main clinical presentations of XP, which include extreme photosensitivity with secondary changes including vesicles, bullae, and conjunctivitis, or disproportionate numbers of lentigines that appear in sun-exposed areas. The onset of these cutaneous manifestations is typically at 1–2 years of age [24]. Chronic cutaneous changes include severe xerosis and premature aging of the skin, with telangiectasias, atrophy, hypopigmentation, and hyperpigmentation. Furthermore, these patients have an increased risk of both NMSCs and melanoma in sun-exposed areas, especially in the head and neck region. These cancers can have an aggressive course, with a tendency to grow rapidly and metastasize early [25]. The median age for NMSC diagnosis is 8 years and median age for melanoma diagnosis is 19–22 years [26, 27]. The overall risk of NMSC is 10,000× normal, and the risk of melanoma is 2,000× normal in patients with XP [27]. Ocular tissue involvement also occurs in almost half of all these patients, resulting in changes like conjunctivitis, ectropion, corneal opacities, and neoplasms [24]. Important extracutaneous findings include progressive neuronal deterioration, which occurs in 20–25% of patients [24, 27]. In the past, diagnosis was made on the basis of clinical features and observations of defective DNA repair, but there is increased utilization of genetic testing, including whole exome sequencing [28].
Management of these patients requires rigorous photoprotection, both when the patient is indoors and outdoors. Measures like UV-resistant films on all windows in a patient’s home and school environments and protective headgear, gloves, and clothing to cover all body surfaces when going outdoors are imperative, given the extraordinary risk of skin cancer in XP patients. High-dose oral isotretinoin has a chemoprophylactic effect and significantly reduces the rate of skin cancer formation during treatment, but the skin cancer formation rate increases again to the pretreatment rate once the medication is stopped [29]. Novel treatments, including a bacterial DNA repair enzyme delivered in a liposomal lotion and gene therapy, are currently under investigation [30]. Regular visits to the dermatologist and ophthalmologist for cancer surveillance are the standard of care. The most common cause of death in XP patients is skin cancer, followed by neurologic decline and internal cancers.
Rothmund-Thomson
Rothmund-Thomson (RT) is another exceedingly rare autosomal recessively inherited genodermatosis. There are about 300 cases of RT reported in the literature [31]. RT1 is a subset without a known causative gene, while RT2 has been linked to a mutation in RECQ4 and an increased risk for malignancy [32]. Mutations in the DNA helicase RecQ family of genes are responsible for Bloom, Werner, and RT, which have different clinical presentations, but share similar mutations that cause gene instability, resulting in a strong predisposition to malignancy [14]. The cutaneous findings usually manifest between 3 and 6 months of life and almost always before the first year of life, with erythema, edema, and blistering, on the cheeks and face, before spreading to the extremities and buttocks [33]. These skin changes can occur in response to ultraviolet light, but can also occur in the setting of minimal sun exposure [34]. This rash typically spares the trunk and abdomen. Chronic changes include atrophy, hyperpigmentation, hypopigmentation, and reticular telangiectasias that can persist throughout the patient’s lifetime. Other cutaneous findings that would support the diagnosis of RT include diffuse hypotrichosis on the scalp and eyebrows, nail dystrophy, and palmoplantar keratosis. Squamous cell carcinoma is the most common cutaneous tumor and represents an important manifestation of RT. Important extracutaneous clues to the diagnosis include cataracts, short stature, and skeletal abnormalities.
The diagnosis of RT is primarily based on characteristic clinical findings, as well as genetic testing for RECQL4 mutations , and treatment for these patients is mainly supportive. In terms of long-term management, it is important to know that patients with RT have increased susceptibility for both osteosarcoma (most common malignancy) and cutaneous SCC. Given these associations, there should be an emphasis on photoprotection and a low threshold for work-up of musculoskeletal complaints. NMSC, especially SCC, presents at a mean age of 34 years, but surveillance with complete skin examinations should begin in adolescence [35].
Bloom Syndrome
Bloom syndrome is an autosomal recessive disease found frequently in the Ashkenazi Jewish population and associated with a mutation in the BLM gene at chromosome 15q26.1 that encodes RECQL3 helicase [36, 37]. The DNA in lymphocytes, which lack this important helicase in affected patients, demonstrate an increase in sister chromatid exchanges and therefore DNA instability [38]. Several hundred cases have been reported in the literature. Erythema of the cheeks in a butterfly distribution after sun exposure is often the earliest cutaneous manifestation appearing within the first few weeks of life. The rash can spread over the face, but tends to spare the extremities and trunk. Photosensitivity resulting in blistering, erythema, and bleeding is commonly seen in any region exposed to UV radiation [36]. These cutaneous changes evolve into chronic changes that include mottled hypo- and hyperpigmentation, telangiectasias, atrophy, and scarring. Other important cutaneous manifestations are café au lait macules and hypopigmented macules, especially over the dorsum of the hands and forearms [39]. The most distinctive extracutaneous feature of Bloom syndrome is proportionate small stature. Other suggestive extracutaneous findings include a characteristic birdlike appearance due to lack of subcutaneous fat, recurrent bacterial infections with associated hypogammaglobulinemia, and impaired fertility in men and women. Diagnosis is ultimately made through karyotype or genetic mutation analysis [40].
The range of malignancies associated with Bloom syndrome is similar to the spectrum seen in the general population, but affects patients with an increased frequency and at a younger age. In the first decade, rare cases of Wilms tumor and osteosarcoma have been reported. In the second decade, hematologic malignancies and skin cancer become more common, and the risk of all other carcinomas, especially colon and breast cancer, increases thereafter [41]. Of note, pulmonary complications like bronchiectasis, while not malignant processes, contribute significantly to the mortality rate and are the second-leading cause of death after malignancy in these patients [41]. Dermatologic care should include photoprotection and frequent monitoring to detect skin cancers. Management can be very difficult because Bloom syndrome patients are susceptible to such a wide spectrum of malignancies, but avoidance of unnecessary radiation and early screening for the common malignancies like breast and colon cancer have been recommended [42].
Cell Matrix Instability
Kindler Syndrome
Kindler syndrome , or congenital bullous poikiloderma , is a rare subtype of epidermolysis bullosa with an autosomal recessive inheritance pattern. It is caused by a mutation in the FERMT1 gene encoding kindlin-1, resulting in dysfunctional actin cytoskeleton-extracellular matrix interaction at multiple intra- and subepidermal levels [43]. To date, there have only been 250 cases of this blistering disease reported [44]. Patients typically present at birth with severe skin fragility and new blisters forming after exposure to trauma or sunlight. Chronic changes including sclerosing features and diffuse cutaneous atrophy affecting the dorsal aspects of the hands (Fig. 5.3) increase with age [45]. Other cutaneous findings include webbing of hands and feet, hyperkeratosis of palms and soles, nail dystrophy, ectropion, gingivitis, and periodontitis leading to premature loss of teeth. Furthermore, patients develop actinic keratoses or SCCs in the third and fourth decade of life, especially on the acral surfaces and oral mucosa, most likely due to ultraviolet-induced DNA damage and chronic inflammation [45]. Extracutaneous findings include esophageal, urethral, and vaginal stenosis and colitis secondary to mucosal inflammation [46]. Given the overlap with other blistering disorders, the gold standard of diagnosis is molecular genetic mutation testing for loss of function mutations in FERMT1 and supportive histological and immunofluorescence studies.
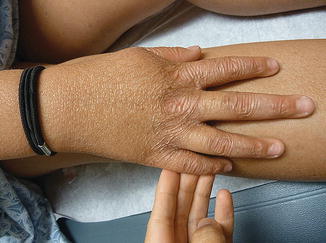
Fig. 5.3
Diffuse cutaneous atrophy of the dorsal aspect of the right hand, consistent with chronic skin changes in Kindler syndrome
In terms of treatment, a multidisciplinary team is essential to minimize complications from strictures and scarring that can affect all mucosal surfaces. Regular dental care is necessary to monitor for and treat the gingivitis. Patients with severe dysphagia secondary to esophageal strictures may require repeat dilatations of the esophagus. Patients with severe colitis, urethral, or vaginal strictures may require surgical intervention. Photoprotection and proper wound care are the mainstays of dermatologic care. Monitoring for skin cancer with regular skin exams at least every 6–12 months, with careful attention to the oral mucosa and areas of chronic ulceration or leukoplakia, should begin at the age of 20 years [47].
Oculocutaneous Albinism
Oculocutaneous albinism (OCA) is a disease entity with autosomal recessive transmission that is caused by a defect in melanin biosynthesis and affects about 1 in 17,000 individuals in the United States [48]. There are at least seven subtypes of OCA, but OCA1 and OCA2 are the most common forms.
The clinical presentation of OCA includes hypopigmentation of the skin, hair, and eyes due to an absence or reduction in melanin biosynthesis, despite normal number of melanocytes. Reduced melanin biosynthesis results in increased sensitivity to UV radiation and a propensity for cutaneous malignancies, especially SCC and BCC. Patients develop signs of early actinic damage, as well as amelanotic or pigmented melanocytic nevi. Cutaneous malignancies develop as early as childhood and present at a mean age of the third and fourth decade of life [49]. The true incidence of melanoma in this population is controversial, but appears to be low [50, 51]. The lack of ocular pigmentation is associated with abnormal optic projections of fibers from the retina to the optic cortex. Patients present with translucency of the iris, reduced visual acuity, photophobia, nystagmus, and strabismus. Treatment should include sun avoidance and ophthalmologic care.
Genodermatoses Associated with Other Causes of Secondary Cutaneous Malignancies
Introduction
In the next section, we will discuss a somewhat heterogeneous group of genodermatoses that are not associated with photosensitivity but predispose to development of SCC, this time in the setting of chronic inflammation and/or scarring. While SCCs tend to occur slightly later in life in this context, we will discuss several genodermatoses that are associated with development of skin cancer during childhood, including epidermolysis bullosa, incontinentia pigmenti, and epidermodysplasia verruciformis.
Key Points
In certain subtypes of epidermolysis bullosa, chronic inflammation, ulceration, and scarring predispose to the development of SCC that may behave aggressively.
Incontinentia pigmenti is characterized by skin changes in a Blaschkoid distribution and is associated with an increased risk for SCC within affected areas.Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree