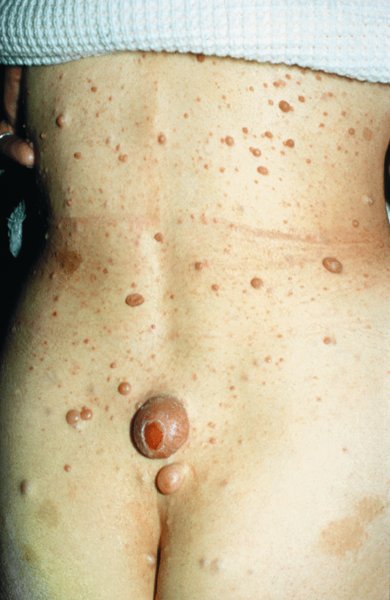
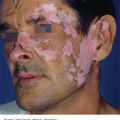
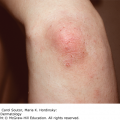
24 The human genome consists of 23 pairs of chromosomes carrying an estimated 30 000 genes. The pairs of matching chromosomes as seen at colchicine-arrested metaphase are numbered in accordance with their size. A centromere divides each chromosome into a shorter (p) and a longer (q) arm. Any individual’s chromosomal makeup (karyotype) can be expressed as their total number of chromosomes plus their sex chromosome constitution. A normal male therefore is 46XY. A shorthand notation exists for recording other abnormalities such as chromosome translocations and deletions. The precise location of any gene can be given by naming the chromosome, the arm of the chromosome (p or q), and the numbers of the band and subband of the chromosome, as seen with Giemsa staining, on which it lies. Filaggrin, one of the genes important for atopic eczema, for instance, lies on chromosome 1q21.3, on the long arm of chromosome 1 at band 21, subband 3. Over the last decade enormous progress has been made in the fields of molecular biology and genetic discovery. Pivotal to these advances was the completion of the Human Genome Project in 2003, which provided a publically available reference template of the human genome. Building on this success, collaborations such as the International HapMap Project mapped common genetic variants (single nucleotide polymorphisms; SNPs) within the genome. Fundamental to these projects was DNA sequencing; initially, using the Sanger sequencing method first described in 1977 but now largely superseded by next generation sequencing (NGS). In the Sanger method one gene is sequenced at a time in contrast to NGS where a DNA sample is fragmented into multiple smaller segments (DNA library), which are sequenced simultaneously (massively parallel sequencing). There are many commercially available NGS platforms which all rely on the same basic steps: DNA extraction; fragmentation in to libraries; amplification; parallel sequencing and imaging; alignment of the sequenced fragments (to a known reference template or de novo alignment) and bioinformatics data analysis. With the advent of NGS, sequencing has become faster, cheaper and universally accessible; a whole genome can now be sequenced in a day. The process can be further streamlined by limiting sequencing to the 1% of the genome that codes for proteins (whole-exome sequencing). However, whole-genome sequencing may be needed to identify non-coding regulatory regions within the introns. The use of NGS has numerous applications: identification of SNPs; assessment of structural variation within segments of DNA such as deletions, duplications, inversions and translocations (copy number variants or CNVs); and detection of novel mutations. Sequencing panels have also been developed to rapidly identify pathological mutations in single genes or to highlight common mutation hotspots in larger sections of the genome. This more targeted approach further aids in the diagnosis of genetic conditions. Sequencing may also be used in combination with established methods of gene identification. Genes are linked if they lie close together on the same chromosome; they will then be inherited together. The closer together they are, the less is the chance of their being separated by cross-overs, one to six of which, depending on length, occur on each chromosome at meiosis. Each member of an affected family has to be examined both for the presence of the trait to be mapped, and also for a marker, usually a DNA probe, which has already been mapped. If linkage is established then the two loci will be close on the same chromosome. The probability of the results of such a study representing true linkage can be expressed as a logarithm of the odds (Lod) score. A score of three or more suggests that the linkage is likely to be genuine. Large populations with a certain phenotype are scanned for the presence of SNPs. These phenotypes are then compared with a reference database to ascertain if any particular SNPs are associated with that particular phenotype. If they are, then sequencing of the section of genome marked by the SNPs may identify disease-causing genes. The technique is especially useful in investigating common conditions with complex modes of inheritance such as psoriasis and eczema. A cloned sequence of DNA, if made single-stranded by heat, will stick to its complementary sequence on a chromosome. Radioactive or fluorescent labelling can be used to indicate its position there. This is used to study the expression of multiple genes simultaneously in a given biological tissue. It again relies on the principle of complementary hybridization. Thousands of cDNA or oligonucleotide probes (a specific known DNA sequence) are bonded to a solid surface (glass slide or silicon chip) at a particular site to form a grid; these may be specific to a particular gene or mutation. mRNA is extracted from the tissue under investigation, reverse transcribed into cDNA and labelled with a fluorescent marker. This is then incubated with the microarray and hybridization occurs. The intensity of fluorescence corresponds to the strength of hybridization and hence to the expression of the gene or sequence under investigation. Complex statistical software is used to analyse the results. This technology has been used to identify the over- or under-expression of genes, the frequency of gene mutations and the occurrence of SNPs in many malignant and inflammatory skin conditions. It should be remembered that just because a gene is over- or under-expressed this might not necessarily correspond to its expression at a protein level. Recently, multiple arrays have been combined in high-throughput gene expression profiling platforms. These have enabled the screening of multiple samples at once, dramatically simplifying the screening of larger clinical populations. Traditional genetics has also been extended by the introduction of several new non-Mendelian concepts of importance in dermatology. Gene expression may be altered even when the genetic code (DNA sequence) remains unchanged; rarely, these changes may be inherited from one generation to the next. The study of these phenomena is termed epigenetics. There are three broad epigenetic mechanisms: (i) DNA hypermethylation tending to supress gene transcription; (ii) modification of histone proteins (proteins tightly associated with DNA to form chromatin), with acetylation being the most studied usually resulting in activation of genes; (iii) the presence of microRNA genes which themselves do not code for a protein but affect the transcription of other genes. Epigenetic changes are thought to have a role in the pathogenesis of multiple inflammatory and malignant skin conditions. Two other important genetic concepts are clinical heterogeneity (when clinically distinct phenotypes are produced by different mutations within the same gene, such as red hair variants caused by different polymorphisms of the MC1R gene) and genetic heterogeneity (when the same clinical picture can be produced by mutations in different genes, as in tuberous sclerosis). Inheritance is important in many of the conditions discussed in other chapters and this has been highlighted in the sections on aetiology. This chapter includes some genetic disorders not covered elsewhere. Red hair is not, of course, a disease but it is the first normal variation in human appearance for which a causative genetic polymorphism was found. The melanocortin-1 receptor (MC1R) gene is found at 16q24.3. While at least 30 genetic variants have been described, three in particular are associated with an increased phaeo- (red) to eu- (black) melanin ratio. Individuals homozygous or heterozygous for one or two of these ‘red hair alleles’ are likely to have pale skin, red hair and poor tanning ability. These relatively common disorders affect about 1 in 3500 people and are inherited as an autosomal dominant trait. There are two main types: von Recklinghausen’s neurofibromatosis (NF1; which accounts for 85% of all cases) and bilateral acoustic neurofibromatosis (NF2); these are phenotypically and genetically distinct. The NF1 gene has been localized to chromosome 17q11.2. It is unusually large (300 kb) and many different mutations within it have now been identified. The NF1 gene is a tumour suppressor gene, the product of which, neurofibromin, interacts with the product of the RAS proto-oncogene. This may explain the susceptibility of NF1 patients to a variety of tumours. The inheritance of NF1 is as an autosomal dominant trait but about half of index cases have no preceding family history. Recently, microRNA expression has been implicated in the control of the NF1 gene. The physical signs include the following: Figure 24.1 Neurofibromatosis: one large but benign neurofibroma has ulcerated over the sacrum. Several café au lait patches are visible.
Other Genetic Disorders
Linkage analysis.
Genome-wide association studies (GWASs).
In situ hybridization.
DNA microarray analysis.
Non-Mendelian genetics
Red hair
The neurofibromatoses
NF1
Cause
Clinical features
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


