32 Orbital hypertelorism
Synopsis









Basic science and disease process
If the frontonasal prominence remains in its embryonic position, the optic placodes cannot migrate toward the midline, and this results in orbital hypertelorism, often associated with various anomalies of the forehead and nose. It had been said that “the face predicts the brain,” and the importance of the centrofacial anomaly appears to parallel the forebrain defect.1 Conversely, interrupted development of the medial prominence may lead to an absence of midline structures with excessive narrowing, such as cyclopia or ethmocephaly. Hypertelorism alone is a mild degree of this sequence at the orbital level.
Radiation, infection, and maternal metabolic imbalances, which can be involved in cleft lip, have not been reported to be responsible for craniofacial malformations. Drugs and chemicals such as tretinoin, thalidomide, corticosteroids, and even aspirin are known to be responsible for malformations; and, while facial development occurs early in pregnancy,2 mothers may ingest various drugs while unaware of being pregnant. Causal environmental and hereditary factors probably play varying epigenetic roles in the formation of particular malformations.
Diagnosis of hypertelorism and patient presentation in facial clefts
Median facial clefts
Midline tissue deficiency malformations are almost always linked with a forebrain deficiency. The term “arhinencephaly” has been used, but holoprosencephaly, a name proposed by De Myer et al.,1 better reflects the lack of median tissues. On the basis of this brain–facial linkage, and including some concepts of Cohen and associates, the holoprosencephalic malformations present with hypotelorism, as opposed to hypertelorism.3
In contrast with the previous group, near-normal to excess tissue midline disorders do not have a high correlation between the facial anomalies and the underlying brain. The deformities can range from a notch in the upper lip and a widened nose to the most severe form of midline cleft. The term “frontonasal dysplasia,” suggested by Sedano et al. for this group of anomalies,4 is used widely, especially amongst geneticists. Frontonasal dysplasia and holoprosencephaly are therefore at opposite ends in this system of classification of midline anomalies.
Tessier classification of facial clefts
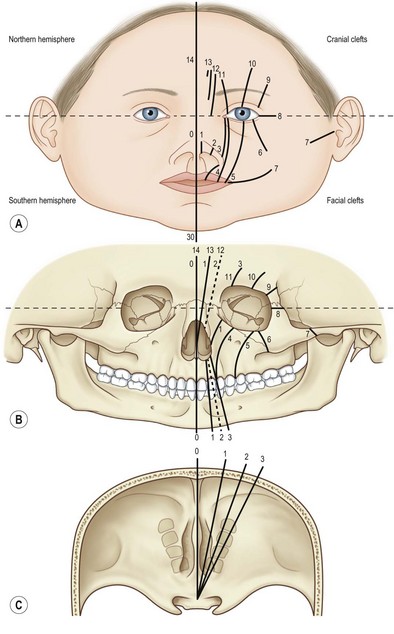
In the Tessier classification,5 the orbit is the reference landmark, common to both the cranium and the face. The clefts, numbered from 0 to 14, rotate around the orbit and follow constant lines through the skeleton and soft tissues (Fig. 32.1). Clefts can be mostly cranial if they run upward from the eyelid (7–14) or mostly facial if they run downward from the palpebral fissure (0–6). They are craniofacial if the upper and lower pathways are connected.
Diagnosis of hypertelorism and patient presentation in faciocraniosynostosis
Hypertelorism is not the main feature of faciocraniosynostosis, and is variable in its presentation.6 When mild, it is frequently left untreated; however, when more noticeable, it can be addressed through a separate surgical step. Depending on the dental occlusion, and particularly the horizontal position of the upper maxillary arch, the treatment of hypertelorism will vary between an orbital shift and a facial bipartition. The timing of treatment is variable according to the pathology; however, in the authors’ experience, skull expansion for craniosynostosis treatment should precede hypertelorism correction.7,8
Crouzon’s syndrome
Described by Crouzon in 1912,9 this syndrome involves only the face and cranium and is not associated with other anomalies elsewhere on the limbs or trunk. The fundamental dysmorphology is an underdeveloped midface presenting with exorbitism secondary to insufficient depth of the orbits, hypoplasia of the malar bones, and a class III malocclusion. Hypertelorism may be present but is usually mild. The nose is short. Brachycephaly is usually present; however, scaphocephaly, plagiocephaly, or even a cloverleaf skull may be present. With an extended classification, the association of facial retrusion and craniosynostosis might be named as a related Crouzon’s anomaly. Commonly, the diagnosis of Crouzon’s syndrome is difficult to make during the first year of life, even if brachycephaly is obvious. It is often difficult to know whether the midface will be affected, even on radiological examination. Midface retrusion and exorbitism appear later in life. In some cases, however, the diagnosis is evident at birth.
The strategy of treatment of faciocraniosynostosis is either two-staged10–12 (fronto-orbital first, then facial advancement later) or a frontofacial monobloc advancement,13–15 preferably with distraction.16 Whenever present, the hypertelorism might be corrected around 4–5 years of age, usually with an orbital shift (Fig. 32.2), because in Crouzon’s disease, the maxillary arch is normal (at least in the horizontal dimension).
Apert’s syndrome (acrocephalosyndactyly)
First described by Apert in 1906,17 this syndrome is easy to recognize because of the associated syndactylies of the hands and feet. The severity of syndactyly is scored according to Cohen and Kreiborg.18 Type 1 is syndactyly of the central three digits, type 2 is syndactyly of digits 2–5, and type 3 is syndactyly of all five digits.
The craniofacial involvement is always marked at birth, in contrast with that in Crouzon’s, with brachycephaly (sometimes asymmetrical) associated with facial retrusion. Both coronal sutures are fused, although some rare cases present with unilateral coronal synostosis or without any synostoses (4 cases without synostoses in our series). The hypertelorism and the anterior open bite add to the distinction from Crouzon’s disease. In fact, the maxillary alveolar arch is higher than the posterior part of the palate. Another main difference with Crouzon’s is the frequent sagittal dehiscence of the sutural system with the forehead frequently remaining wide open during the first year of life; this contributes to the abnormally wide forehead and face in this syndrome. Associated abnormalities of the central nervous system are more frequent than in other syndromic craniosynostoses. The exception is Chiari-like malformations, which are common in Crouzon’s, but, due to premature fusions of the lambdoid sutures, are uncommon in Apert’s.19 Because of these patients’ divergent frontal plane, bipartition procedures are more adapted to correct Apert’s-associated hypertelorism. Nevertheless, skull expansion (either anterior or posterior) is performed first, usually before 1 year of age to address the craniosynostotic skull.
Patient selection
Orbital shift or bipartition?
The choice between orbital shift and bipartition is mainly linked to a series of factors:
• The maxillary arch: if the maxillary arch is narrow and inverted with the incisors being higher than the molars, bipartition is the operation of choice because it widens the maxilla and improves the angle of the upper dentition. On the other hand, if the maxillary arch and occlusion are normal, it is preferable to avoid an interpterygomaxillary disjunction.
• The axis of the orbits: if the axis is normal, a horizontal mobilization is satisfactory; if the orbits are laterally and downwardly oblique, bipartition is required.
• The nasal fossae: if they are narrow, bipartition and medialization of the upper face improve the airways by enlarging the lower part of the face.
• Extent of the hypertelorism: bipartition is the operation of choice in the most severe cases, whereas orbital shift is indicated for more limited displacements.
• The bipartition procedure can also be used to get access to a lesion of the cranial base. Some midline clefts are associated with an encephalocele of the ethmoidosphenoidal area. After midline splitting, easy access to the encephalocele can be obtained.
Treatment/surgical technique
Surgical principles in facial clefts
Tessier’s breakthrough collaboration between plastic and neurological surgery allowed him to conceptually minimize the surgical border existing between the face and the skull and dramatically enhance the surgical treatment of upper facial clefts.20 Tessier demonstrated that the frontocranial route was a good approach to access the nose and orbits and that frontocranial problems can be treated simultaneously with facial problems. The fear of contamination from the facial cavities was so great among neurosurgeons in 1967 that, for their first case, Tessier and his neurosurgical colleague, Guyot, placed a dermal skin graft on the dura of the anterior cranial fossa after it had been elevated, voluntarily sacrificing both the olfactory nerves. A few months later, they performed the combined craniofacial approach. By 1970, a one-stage procedure to address the orbits from superior and inferior approaches was considered safe, and the olfactory nerves could be preserved (Converse).21 Preliminary disinfection of the nasal cavities, dissection of the mucosal domes and their immediate repair if opened, preservation or perfect repair of dura, changing of instruments if passing through the facial cavities, and perioperative antibiotic therapy were preventive measures that helped to avoid infection, osteitis, and meningitis.
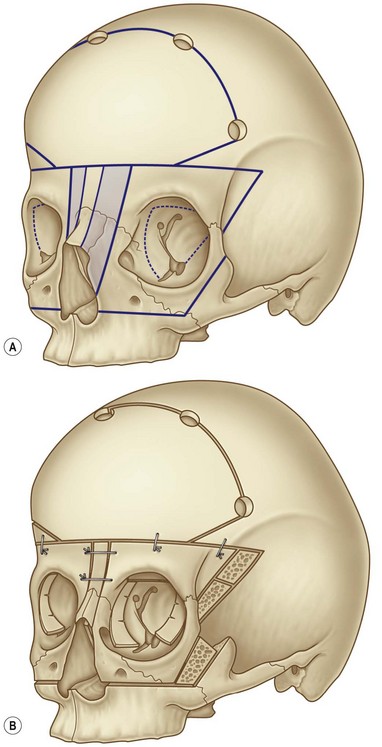
The classic approach described by Tessier et al. in 196720 consisted, after removal of the enlarged medial portion, of an en bloc mobilization of the orbits medially, the lower horizontal cuts being situated below the infraorbital rim, through the malar bone and the maxilla. In Tessier’s protocol, especially in adult patients, a supraorbital bar is kept in place in order to ensure stability of the mobilized orbits.22
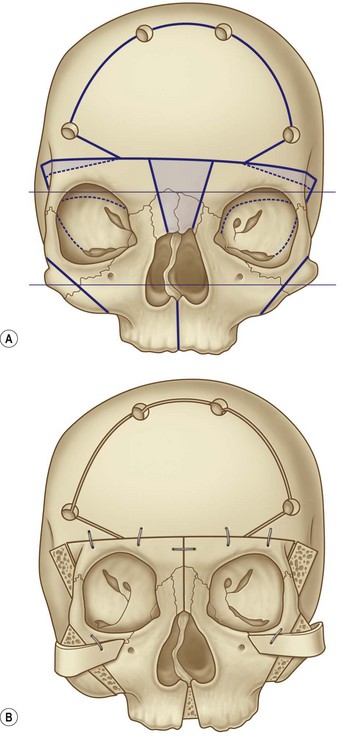
Bipartition, proposed by van der Meulen23,24 and developed by Tessier,25,26 represents a mobilization of the two hemifaces (Fig. 32.3). Instead of cutting below the orbits, the surgeon makes the osteotomies through the zygomatic arch, the pterygomaxillary junction and medially through the palate. The medial resection must have a “V” shape because there will be an element of rotation, along with a narrowing on the midline. The rotation allows for widening of the maxillary arch and the nasal fossae and also for changing the axis of the orbits, which have a lateral slant.
Surgical techniques in facial clefts
Principles
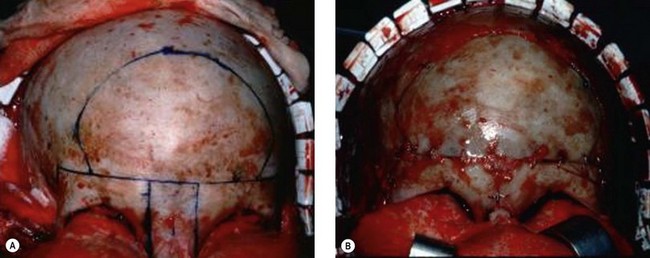
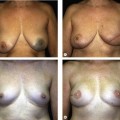
The two main aims of surgery for this condition are to bring the orbits closer together and to create a nose of normal appearance. The basic anatomical anomaly is an increase in both the interorbital distance and the nasal bones whilst the glabellar region is much wider than normal. The enlarged portion of the midline structures is removed and, after mobilization of the orbits, the nasal skeleton is adjusted with the help of a bone graft if more dorsal projection is needed (Fig. 32.4).
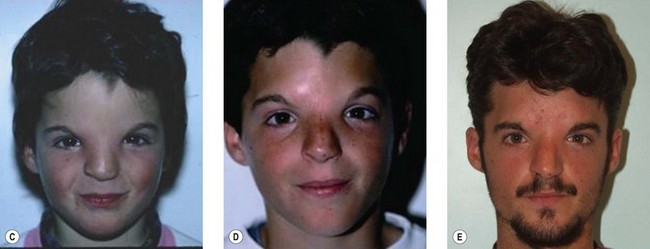
Not all hypertelorisms are symmetrical, and asymmetrical cases are more complex to correct. The cranial base is also asymmetrical and so the different distortions must be carefully evaluated by computed tomography three-dimensional reconstructions. Sometimes both orbits have to be moved in different directions.26,27
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


