45–51 °C
within minutes
51 and 70 °C
within seconds
Above 70 °C
less than a second
The depth and severity of the burn are also determined by the ability of the contact material to transfer heat, a factor referred to as the specific heat. This is especially important in scald and contact burns. The knowledge about the material type allows for a more accurate estimate of tissue damage.
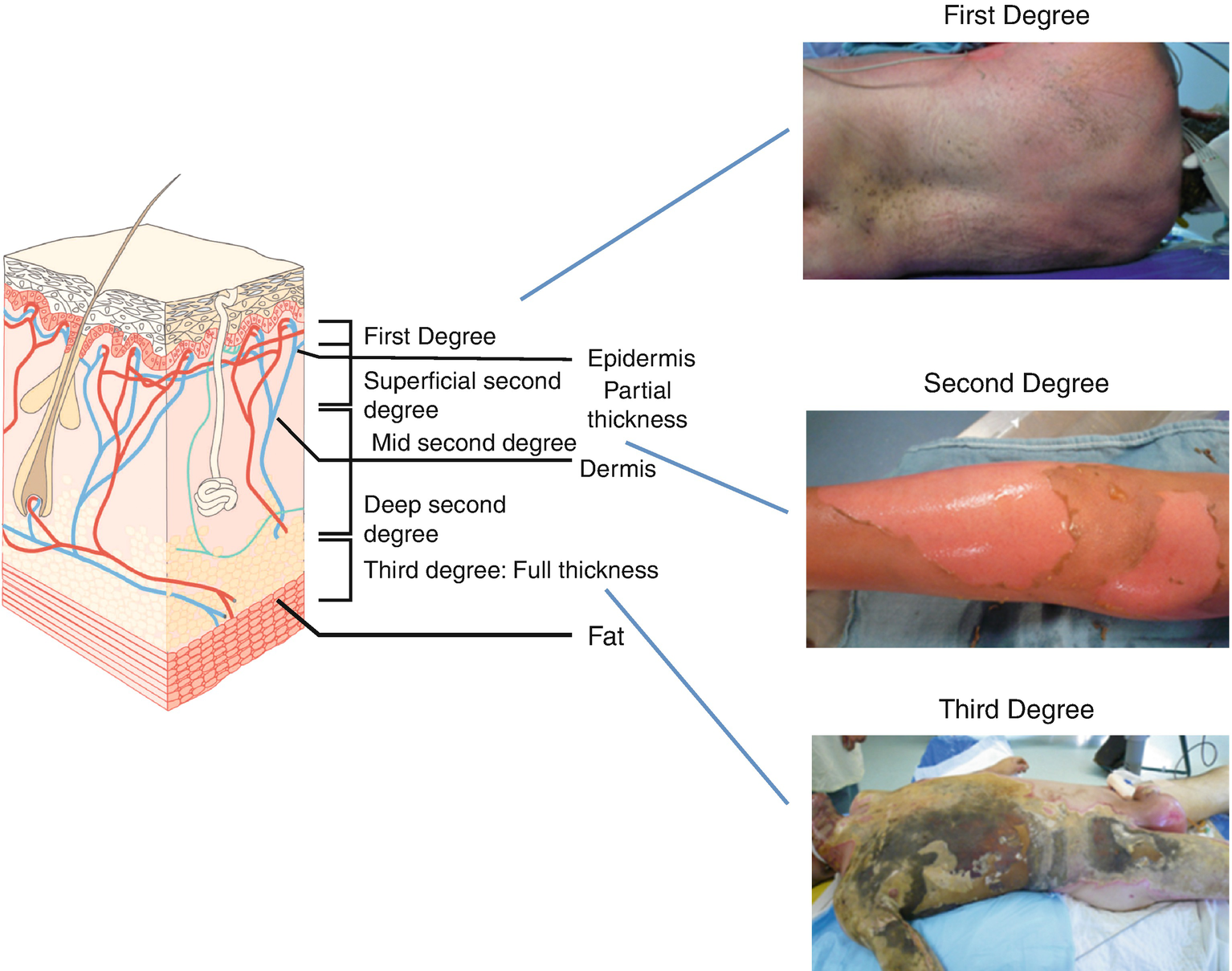
Definition: Burn depth is determined by the time of exposure, the temperature at which the burn occurred, and the caloric equivalent of the burn media.
Another determinant of the severity of burn is the location of the burn wound and the age of the burn patient. The thickness of the skin layers increases from the age of 5 up to the age of 50. In elderly patients, the thickness starts to decrease at the age of 65. The epidermis can vary by location from 0.03 up to 0.4 mm. The depth of burn varies depending on the degree of tissue damage. Burn depth is classified into degree of injury in the epidermis, dermis, subcutaneous fat, and underlying structures.
- (a)
I degree: Superficial burn of the epidermis
First-degree burns are painful, erythematous, and blanch to the touch with an intact epidermal barrier. Examples include sunburn or a minor scald from a kitchen accident. These burns do not result in scarring, and treatment is aimed at comfort with the use of topical soothing salves with or without aloe and oral nonsteroidal anti-inflammatory agents.
- (b)
IIa degree: Burn including epidermis and superficial dermis
- (c)
IIb degree: Burn including epidermis and deep dermis
Second-degree burns are divided into two types: superficial and deep. All second-degree burns have some degree of dermal damage, by definition, and the division is based on the depth of injury into the dermis. Superficial dermal burns are erythematous, painful, blanch to touch, and often blister. Examples include scald injuries from overheated bathtub water and flash flame burns. These wounds spontaneously re-epithelialize from retained epidermal structures in the rete ridges, hair follicles, and sweat glands in 1–2 weeks. After healing, these burns may have some slight skin discoloration over the long term. Deep dermal burns into the reticular dermis appear more pale and mottled, do not blanch to touch, but remain painful to pinprick. These burns heal in 2–5 weeks by re-epithelialization from hair follicles and sweat gland keratinocytes, often with severe scarring as a result of the loss of dermis.
- (d)
III degree: Burn including epidermis and dermis and subcuticular layer
Third-degree burns are full-thickness through the epidermis and dermis and are characterized by a hard, leathery eschar that is painless and black, white, or cherry red. No epidermal or dermal appendages remain; thus, these wounds must heal by re-epithelialization from the wound edges. Deep dermal and full-thickness burns require excision with skin grafting from the patient to heal the wounds in a timely fashion.
- (e)
IV degree: All dermal layers including fascia, muscles, and/or bones
Fourth-degree burns involve other organs beneath the skin, such as muscle, bone, and brain.
Currently, burn depth is most accurately assessed by judgment of experienced practitioners. Accurate depth determination is critical to wound healing as wounds that will heal with local treatment are treated differently than those requiring operative intervention. Examination of the entire wound by the physicians ultimately responsible for their management then is the gold standard used to guide further treatment decisions. New technologies, such as the multi-sensor laser Doppler flow-meter, hold promise for quantitatively determining burn depth [10].
18.1.2 Burn Size
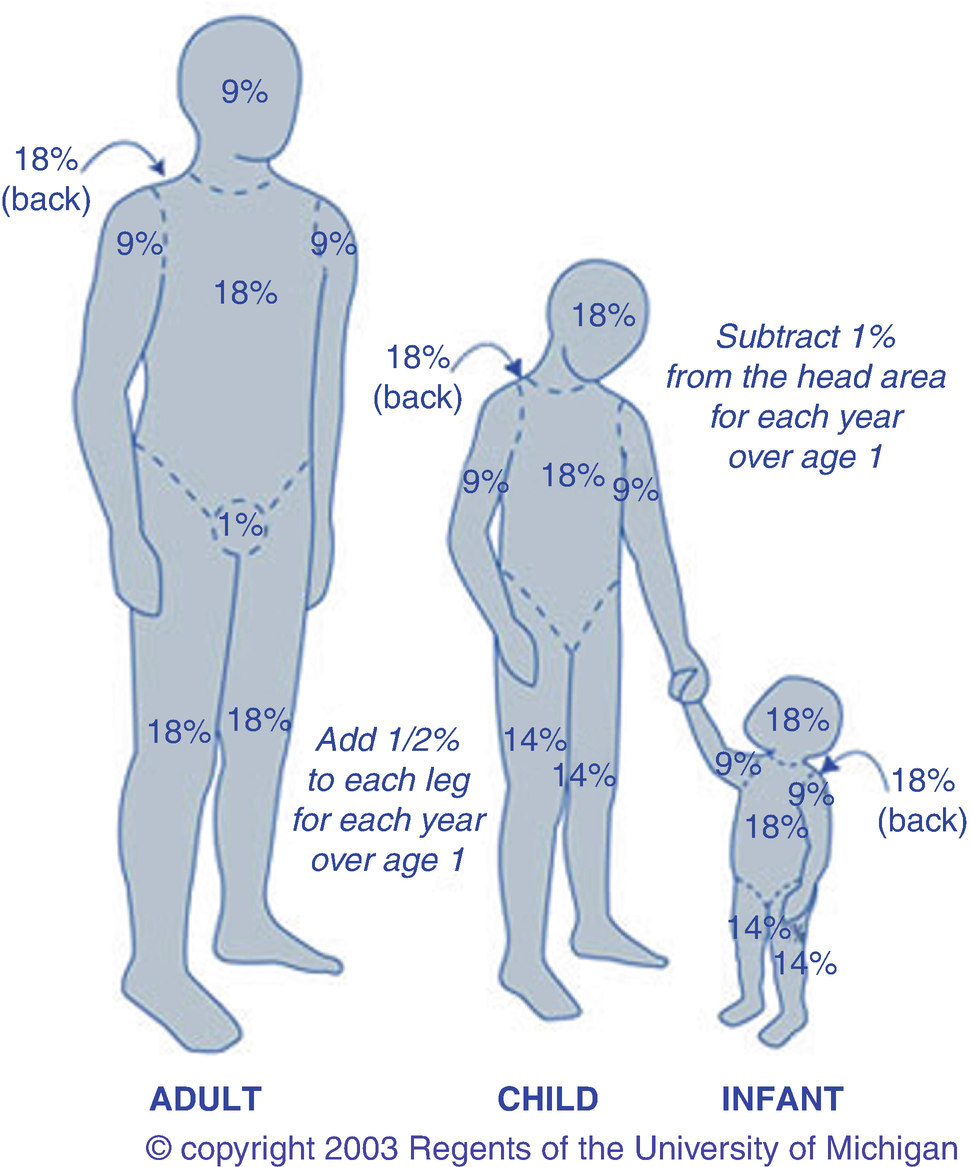
Determination of burn size estimates the extent of injury. Burn size is generally assessed by the “rule of nines.” In adults, each upper extremity and the head and neck are 9% of the TBSA, the lower extremities and the anterior and posterior trunk are 18% each, and the perineum and genitalia are assumed to be 1% of the TBSA. Another method of estimating smaller burns is to equate the area of the open hand (including the palm and the extended fingers) of the patient to be approximately 1% TBSA and then to transpose that measurement visually onto the wound for a determination of its size. This method is crucial when evaluating burns of mixed distribution.
Children have a relatively larger portion of the body surface area in the head and neck, which is compensated for by a relatively smaller surface area in the lower extremities. Infants have 21% of the TBSA in the head and neck and 13% in each leg, which incrementally approaches the adult proportions with increasing age. The Berkow formula is used to accurately determine burn size in children.
18.1.2.1 Etiology
The causes include injury from flame (fire), hot liquids (scald), contact with hot or cold objects, chemical exposure, and/or conduction of electricity. The first three induce cellular damage by the transfer of energy, which induces a coagulation necrosis. Chemical burns and electrical burns cause direct injury to cellular membranes in addition to the transfer of heat.
18.2 Systemic Changes
The release of cytokines and other inflammatory mediators at the site of injury has a systemic effect once the burn reaches 30% of total body surface area (TBSA). Cutaneous thermal injury greater than one-third of the total body surface area invariably results in the severe and unique derangements of cardiovascular function called burn shock. Shock is an abnormal physiologic state in which tissue perfusion is insufficient to maintain adequate delivery of oxygen and nutrients and removal of cellular waste products. Before the nineteenth century, investigators demonstrated that after a burn, fluid is lost from the blood and blood becomes thicker; and in 1897, saline infusions for severe burns were first advocated [11, 12]. However, a more complete understanding of burn pathophysiology was not reached until the work of Frank Underhill [13]. He found that unresuscitated burn shock correlates with increased hematocrit values in burned patients, which are secondary to fluid and electrolyte loss after burn injury. Increased hematocrit values occurring shortly after severe burn were interpreted as a plasma volume deficit. Cope and Moore showed that the hypovolemia of burn injury resulted from fluid and protein translocation into both burned and non-burned tissues [14].
Over the last 80 years an extensive record of both animal and clinical studies has established the importance of fluid resuscitation for burn shock. Investigations have focused on correcting the rapid and massive fluid sequestration in the burn wound and the resultant hypovolemia. The peer-reviewed literature contains a large experimental and clinical database on the circulatory and microcirculatory alterations associated with burn shock and edema generation in both the burn wound and non-burned tissues. During the last 40 years, research has focused on identifying and defining the release mechanisms and effects of the many inflammatory mediators produced and released after burn injury [15].
It is now well recognized that burn shock is a complex process of circulatory and microcirculatory dysfunction that is not easily or fully repaired by fluid resuscitation. Severe burn injury results in significant hypovolemic shock and substantial tissue trauma, both of which cause the formation and release of many local and systemic mediators [16–18]. Burn shock results from the interplay of hypovolemia and the release of multiple mediators of inflammation with effects on both the microcirculation as well as the function of the heart, large vessels, and lungs. Subsequently, burn shock continues as a significant pathophysiologic state, even if hypovolemia is corrected. Increases in pulmonary and systemic vascular resistance (SVR) and myocardial depression occur despite adequate preload and volume support [18–22]. Such cardiovascular dysfunctions can further exacerbate the whole body inflammatory response into a vicious cycle of accelerating organ dysfunction [17, 18, 23]. Hemorrhagic hypovolemia with severe mechanical trauma can provoke a similar form of shock.
18.2.1 Hypovolemia and Rapid Edema Formation
Burn injury causes extravasation of plasma into the burn wound and the surrounding tissues. Extensive burn injuries are hypovolemic in nature and characterized by the hemodynamic changes similar to those that occur after hemorrhage, including decreased plasma volume, cardiac output, urine output, and an increased systemic vascular resistance with resultant reduced peripheral blood flow [16, 18, 24–26]. However, as opposed to a fall in hematocrit with hemorrhagic hypovolemia due to transcapillary refill an increase in hematocrit and hemoglobin concentration will often appear even with adequate fluid resuscitation. As in the treatment of other forms of hypovolemic shock, the primary initial therapeutic goal is to quickly restore vascular volume and to preserve tissue perfusion in order to minimize tissue ischemia. In extensive burns (>25%TBSA), fluid resuscitation is complicated not only by the severe burn wound edema, but also by extravasated and sequestered fluid and protein in non-burned soft tissue. Large volumes of resuscitation solutions are required to maintain vascular volume during the first several hours after an extensive burn. Data suggests that despite fluid resuscitation normal blood volume is not restored until 24–36 h after large burns [27].
Edema develops when the rate by which fluid is filtered out of the microvessels exceeds the flow in the lymph vessels draining the same tissue mass. Edema formation often follows a biphasic pattern. An immediate and rapid increase in the water content of burn tissue is seen in the first hour after burn injury [25, 28]. A second and more gradual increase in fluid flux of both the burned skin and non-burned soft tissue occurs during the first 12–24 h following burn trauma [17, 28]. The amount of edema formation in burned skin depends on the type and extent of injury [25, 29] and whether fluid resuscitation is provided as well as the type and volume of fluid administered [30]. However, fluid resuscitation elevates blood flow and capillary pressure contributing to further fluid extravasation. Without sustained delivery of fluid into the circulation edema fluid is somewhat self-limited as plasma volume and capillary pressure decrease. The edema development in thermal-injured skin is characterized by the extreme rapid onset of tissue water content, which can double within the first hour after burn [25, 31]. Leape and colleagues found a 70–80% water content increase in a full-thickness burn wound 30 min after burn injury with 90% of this change occurring in the first 5 min [26, 32, 33]. There was little increase in burn wound water content after the first hour in the nonresuscitated animals. In resuscitated animals or animals with small wounds, adequate tissue perfusion continues to “feed” the edema for several hours. Demling and others used dichromatic absorptiometry to measure edema development during the first week after an experimental partial-thickness burn injury on one hind limb in sheep [28]. Even though edema was rapid with over 50% occurring in the first hour, maximum water content did not occur until 12–24 h after burn injury.
18.2.2 Altered Cellular Membranes and Cellular Edema
In addition to a loss of capillary endothelial integrity, thermal injury also causes change in the cellular membranes . Baxter found in burns of >30% a systemic decrease in cellular transmembrane potentials as measured in skeletal muscle away from the site of injury [20]. It would be expected that the directly injured cell would have a damaged cell membrane, increasing sodium and potassium fluxes, resulting in cell swelling. However, this process also appears in cells that are not directly heat-injured. Micropuncture techniques have demonstrated partial depolarization in the normal skeletal muscle membrane potential of −90 mV to levels of −70–−80 mV; cell death occurs at −60 mV. The decrease in membrane potentials is associated with an increase in intracellular water and sodium [34–36]. Similar alterations in skeletal membrane functions and cellular edema have been reported in hemorrhagic shock [34, 36] also in the cardiac, liver, and endothelial cells [37–39]. Early investigators of this phenomenon postulated that a decrease in ATP levels or ATPase activity was the mechanism for membrane depolarization, however, other research suggests that it may result from an increased sodium conductance in membranes or an increase in sodium-hydrogen antiport activity [35, 38]. Resuscitation of hemorrhage rapidly restores depolarized membrane potentials to normal, but resuscitation of burn injury only partially restores the membrane potential and intracellular sodium concentrations to normal levels, demonstrating that hypovolemia alone is not totally responsible for the cellular swelling seen in burn shock [40]. A circulating shock factor(s) is likely to be responsible for the membrane depolarization, [41–43] but surprisingly, the molecular characterization of such a circulating factor has not been elucidated; suggesting that it has a complex structure. Data suggests it has a large molecular weight, >80 kDalton [44]. Membrane depolarization may be caused by different factors in different states of shock. Very little is known about the time course of the changes in membrane potential in clinical burns. More importantly, we do not know the extent to which the altered membrane potentials affect total volume requirements and organ function in burn injury or even shock in general.
18.2.3 Mediators of Burn Injury
Many mediators have been proposed to account for the changes in permeability after burn, including histamine, b serotonin, bradykinin, prostaglandins, leukotrienes, platelet-activating factor, and catecholamines, among others [10, 45–49]. The exact mechanism(s) of mediator-induced injury are of considerable clinical importance, as this understanding would allow for the development of pharmacologic modulation of burn edema and shock by mediator inhibition.
Histamine: Histamine is most likely to be the mediator responsible for the early phase of increased microvascular permeability seen immediately after burn. Histamine causes large endothelial gaps to transiently form as a result of the contraction of venular endothelial cells [50]. Histamine is released from mast cells in thermal-injured skin; however, the increase in histamine levels and its actions are only transient. Histamine also can cause the rise in capillary pressure (Pc) by arteriolar dilation and venular contraction. Statistically significant reductions in burn edema have been achieved with histamine blockers and mast cell stabilizers when tested in acute animal models [50]. Friedl et al. demonstrated that the pathogenesis of burn edema in the skin of rats appears to be related to the interaction of histamine with xanthine oxidase and oxygen radicals [51]. Histamine and its metabolic derivatives increased the catalytic activity of xanthine oxidase (but not xanthine dehydrogenase) in rat plasma and in rat pulmonary artery endothelial cells. In thermally injured rats, levels of plasma histamine and xanthine oxidase rose in parallel, in association with the uric acid increase. Burn edema was greatly attenuated by treating rats with the mast cell stabilizer, cromolyn, complement depletion or the H2 receptor antagonist, cimetidine; but was unaffected by neutrophil depletion [52–54]. Despite encouraging results in animals, beneficial antihistamine treatment of human burn injury has not been demonstrated although antihistamines are administered to reduce risk of gastric ulcers.
Prostaglandins: Prostaglandins are potent vasoactive autocoids synthesized from the arachidonic acid released from burned tissue and inflammatory cells and contribute to the inflammatory response of burn injury [55, 56]. Macrophages and neutrophils are activated through the body; infiltrate the wound and release prostaglandin as well as thromboxanes, leukotrienes, and interleukin-1. These wound mediators have both local and systemic effects. Prostaglandin E2 (PGE2) and leukotrienes LB4 and LD4 directly and indirectly increase microvascular permeability [57]. Prostacyclin (PGI2) is produced in burn injury and is also a vasodilator, but also may cause direct increases in capillary permeability. PGE2 appears to be one of the more potent inflammatory prostaglandins, causing the post-burn vasodilation in wounds, which, when coupled with the increased microvascular permeability amplifies edema formation [58, 59].
Thromboxane: Thromboxane A2 (TXA2) and its metabolite, thromboxane B2 (TXB2) are produced locally in the burn wound by platelets [50]. Vasoconstrictor thromboxanes may be less important in edema formation; however, by decreasing blood flow they can contribute to a growing zone of ischemia under the burn wound and can cause the conversion of a partial-thickness wound to a deeper full-thickness wound. The serum level of TXA and TXA2/PGI2 ratios are increased significantly in burn patients [60]. Heggers showed the release of TXB2 at the burn wound was associated with local tissue ischemia, and that thromboxane inhibitors prevented the progressive dermal ischemia associated with thermal injury and thromboxane release [61, 62]. The TXA2 synthesis inhibitor anisodamine also showed beneficial macrocirculatory effects by restoring the hemodynamic and rheological disturbances towards normal. Demling showed that topically applied ibuprofen (which inhibits the synthesis of prostaglandins and thromboxanes) decreases both local edema and prostanoid production in burned tissue without altering systemic production [63]. On the other hand, systemic administration of ibuprofen did not modify early edema, but did attenuate the post-burn vasoconstriction that impaired adequate oxygen delivery to tissue in burned sheep [64]. Although cyclooxgenase inhibitors have been used after burn injury, neither their convincing benefit, nor their routine clinical use has been reported.
Kinins: Bradykinin is a local mediator of inflammation that increases venular permeability. It is likely that bradykinin production is increased after burn injury, but its detection in blood or lymph can be difficult owing to the simultaneous increase in kininase activity and the rapid inactivation of free kinins. The generalized inflammatory response after burn injury favors the release of bradykinin [65]. Pretreatment of burn-injured animals with aprotinin, a general protease inhibitor, should have decreased the release of free kinin, but no effect on edema was noted [66]. On the other hand, pretreatment with a specific bradykinin receptor antagonist reduced edema in full-thickness burn wound in rabbits [8].
Serotonin: Serotonin is released early after burn injury [67]. This agent is a smooth-muscle constrictor of large blood vessels. Antiserotonin agents such as ketanserin have been found to decrease peripheral vascular resistance after burn injury, but have not been reported to decrease edema [67]. On the other hand, the pretreatment effect of methysergide, a serotonin antagonist, reduces hyperemic or increases blood flow response in the burn wounds of rabbits, along with reducing the burn edema [8]. Methysergide did not prevent increases in the capillary reflection coefficient or permeability [68]. Ferrara and colleagues found a dose-dependent reduction of burn edema when methysergide was given preburn to dogs, but claimed that this was not attributable to blunting of the regional vasodilator response [68]. Zhang et al. reported a reduction in nonnutritive skin blood flow after methysergide administration to burned rabbits [69].
Catecholamines: Circulating catecholamines epinephrine and norepinephrine are released in massive amounts after burn injury [17, 70, 71]. On the arteriolar side of the microvessels, these agents cause vasoconstriction via alpha 1 receptor activation, which tends to reduce capillary pressure, particularly when combined with the hypovolemia and the reduced venous pressure of burn shock [50]. Reduced capillary pressure may limit edema and induce interstitial fluid to reabsorb from non-burned skin, skeletal muscle, and visceral organs in nonresuscitated burn shock. Further, catecholamines, via β-agonist activity, may also partially inhibit increased capillary permeability induced by histamine and bradykinin [50]. These potentially beneficial effects of catecholamines may not be operative in directly injured tissue and may also be offset in non-burned tissue by the deleterious vasoconstrictor and ischemic effects. The hemodynamic effects of catecholamines will be discussed later in the chapter.
Oxygen Radicals: Oxygen radicals play an important inflammatory role in all types of shock, including burn. These short-lived elements are highly unstable reactive metabolites of oxygen; each one has an unpaired electron, creating them into strong oxidizing agents [72]. Superoxide anion (O2−), hydrogen peroxide (H2O2), and hydroxyl ion (OH−) are produced and released by activated neutrophils after any inflammatory reaction or reperfusion of ischemic tissue. The hydroxyl ion is believed to be the most potent and damaging of the three. The formation of the hydroxyl radical requires free ferrous iron (Fe2) and H2O2. Evidence that these agents are formed after burn injury is the increased lipid peroxidation found in circulating red blood cells and biopsied tissue [53, 72, 73]. Demling showed that large doses of deferoxamine (DFO), an iron chelator, when used for resuscitation of 40% TBSA in sheep, prevented systemic lipid peroxidation and decreased the vascular leak in non-burned tissue while also increasing oxygen utilization [74]. However, DFO may have accentuated burned tissue edema, possibly by increasing the perfusion of burned tissue.
Nitric oxide (NO) simultaneously generated with the superoxide anion can lead to the formation of peroxynitrite (ONOO−). The presence of nitrotyrosine in burned skin found in the first few hours after injury suggests that peroxynitrite may play a deleterious role in burn edema [75]. On the other hand, the blockade of NO synthase did not reduce burn edema, while treatment with the NO precursor arginine reduces burn edema [76]. NO may be important for maintaining perfusion and limiting the zone of stasis in burned skin [77]. Although the pro- and anti-inflammatory roles of NO remain controversial, it would appear that the acute beneficial effects of NO generation out-weigh any deleterious effect in burn shock.
Antioxidants, namely agents that either directly bind to the oxygen radicals (scavengers) or cause their further metabolism, have been evaluated in several experimental studies [78, 79]. Catalase, which removes H2O2 and superoxide dismutase (SOD), which removes radical O2−, have been reported to decrease the vascular loss of plasma after burn injury in dogs and rats [53, 78].
The plasma of thermally injured rats showed dramatic increases in levels of xanthine oxidase activity, with peak values appearing as early as 15 min after thermal injury. Excision of the burned skin immediately after the thermal injury significantly diminished the increase in plasma xanthine oxidase activity [51, 53]. The skin permeability changes were attenuated by treating the animals with antioxidants (catalase, SOD, dimethyl sulfoxide, dimethylthiourea) or an iron chelator (DFO), thus supporting the role of oxygen radicals in the development of vascular injury as defined by increased vascular permeability [53]. Allopurinol, a xanthine oxidase inhibitor, markedly reduced both burn lymph flow and levels of circulating lipid peroxides, and further prevented all pulmonary lipid peroxidation and inflammation. This suggests that the release of oxidants from burned tissue was in part responsible for local burn edema, as well as distant inflammation and oxidant release [73]. The failure of neutrophil depletion to protect against the vascular permeability changes and the protective effects of the xanthine oxidase inhibitors (allopurinol and lodoxamide tromethamine) suggests that plasma xanthine oxidase is the more likely source of the oxygen radicals involved in the formation of burn edema. These oxygen radicals can increase vascular permeability by damaging microvascular endothelial cells [51, 53]. The use of antioxidants has been extensively investigated in animals, and some clinical trials suggest benefit. Antioxidants (vitamin C and E) are routinely administered to patients at many burn centers. High doses of antioxidant ascorbic acid (vitamin C) have been found to be efficacious in reducing fluid needs in burn-injured experimental animals when administered post-burn [80–82]. The use of high doses (10–20 g per day) of vitamin C was shown to be effective in one clinical trial, but ineffective in another [83, 84]. High dose vitamin C has not received wide clinical usage.
Platelet Aggregation Factor: Platelet aggregation (or activating) factor (PAF) can increase capillary permeability and is released after burn injury [66, 85]. Ono and colleagues showed in scald-injured rabbits that TCV-309 (Takeda Pharmaceutical Co Ltd., Japan), a PAF antagonist, infused soon after burn injury blocked edema formation in the wound and significantly inhibited PAF increase in the damaged tissue in a dose-dependent manner. In contrast, the superoxide dismutase content in the group treated with TCV-309 was significantly higher than that of the control group [85]. These findings suggest that the administration of large doses of a PAF antagonist immediately after injury may reduce burn wound edema and the subsequent degree of burn shock by suppressing PAF and superoxide radical formation.
Angiotensin II and Vasopressin: Angiotensin II and vasopressin or antidiuretic hormone (ADH) are two hormones that participate in the normal regulation of extracellular fluid volume by controlling sodium balance and osmolality through renal function and thirst [50]. However, during burn shock where sympathetic tone is high and volume receptors are stimulated, both hormones can be found in supranormal levels in the blood. Both are potent vasoconstrictors of terminal arterioles with little effect on the venules. Angiotensin II may be responsible for the selective gut and mucosal ischemia, which can cause translocation of endotoxins and bacteria and the development of sepsis and even multi-organ failure [86, 87]. In severely burn-injured patients, angiotensin II levels were elevated two to eight times normal in the first 1–5 days after burn injury with peak levels occurring on day 3 [88]. Vasopressin had peak levels of 50 times normal upon admission and declined towards normal over the first 5 days after burn injury. Vasopressin, along with catecholamines may be largely responsible for increased system vascular resistance and left heart afterload, which can occur in resuscitated burn shock. Sun and others used vasopressin-receptor antagonist in rats with burn shock to improve hemodynamics and survival time, while vasopressin infusion exacerbated burn shock [89].
Corticotropin-releasing Factor: Corticotropin-releasing factor (CRF) has proven to be efficacious in reducing protein extravasation and edema in burned rat paw. CRF may be a powerful natural inhibitory mediator of the acute inflammatory response of the skin to thermal injury [90].
18.2.3.1 Hemodynamic Consequences of Acute Burns
The cause of reduced cardiac output (CO) during the resuscitative phase of burn injury has been the subject of considerable debate. There is an immediate depression of cardiac output before any detectable reduction in plasma volume. The rapidity of this response suggests a neurogenic response to receptors in the thermally injured skin or increased circulating vasoconstrictor mediators. Soon after injury a developing hypovolemia and reduced venous return undeniably contribute to the reduced cardiac output. The subsequent persistence of reduced CO after apparently adequate fluid therapy, as evidenced by a reduction in heart rate and restoration of both arterial blood pressure and urinary output, has been attributed to circulating myocardial depressant factor(s), which possibly originates from the burn wound [21, 22]. Demling and colleagues showed a 15% reduction in CO despite an aggressive volume replacement protocol after a 40% scald burn in sheep [28]. However, there are also sustained increases in catecholamine secretion and elevated systemic vascular resistance for up to 5 days after burn injury [70, 88]. Michie and others measured CO and SVR in anesthetized dogs resuscitated after burn injury [91]. They found that CO fell shortly after injury and then returned towards normal, however, reduced CO did not parallel the blood volume deficit. They concluded that the depression of CO resulted not only from decreased blood volume and venous return, but also from an increased SVR and from the presence of a circulating myocardial depressant substance. Thus, there are multiple factors that can significantly reduce CO after burn injury. However, resuscitated patients suffering major burn injury can also have supranormal CO from 2 to 6 days post-injury. This is secondary to the establishment of a hypermetabolic state.
18.2.3.2 Hypermetabolic Response to Burn Injury
Marked and sustained increases in catecholamine, glucocorticoid, glucagon, and dopamine secretion are thought to initiate the cascade of events leading to the acute hypermetabolic response with its ensuing catabolic state [92–100]. The cause of this complex response is not well understood. However, interleukins 1 and 6, platelet-activating factor, tumor necrosis factor (TNF), endotoxin, neutrophil-adherence complexes, reactive oxygen species, nitric oxide, and coagulation as well as complement cascades have also been implicated in regulating this response to burn injury [101]. Once these cascades are initiated, their mediators and by-products appear to stimulate the persistent and increased metabolic rate associated with altered glucose metabolism seen after severe burn injury [102].
Several studies have indicated that these metabolic phenomena post-burn occur in a timely manner, suggesting two distinct pattern of metabolic regulation following injury [103]. The first phase occurs within the first 48 h of injury and has classically been called the “ebb phase” [103, 104], characterized by decreases in cardiac output, oxygen consumption, and metabolic rate as well as impaired glucose tolerance associated with its hyperglycemic state. These metabolic variables gradually increase within the first 5 days post-injury to a plateau phase (called the “flow” phase), characteristically associated with hyperdynamic circulation and the above-mentioned hypermetabolic state. Insulin release during this time period was found to be twice that of controls in response to glucose load [105, 106] and plasma glucose levels are markedly elevated, indicating the development of an insulin resistance [106, 107]. Current understanding has been that these metabolic alterations resolve soon after complete wound closure. However, recent studies found that the hypermetabolic response to burn injury may last for more than 12 months after the initial event [92, 93, 100, 108]. We found in recent studies that sustained hypermetabolic alterations post-burn, indicated by persistent elevations of total urine cortisol levels, serum cytokines, catecholamines, and basal energy requirements, were accompanied by impaired glucose metabolism and insulin sensitivity that persisted for up to 3 years after the initial burn injury [109].
A 10–50-fold elevation of plasma catecholamines and corticosteroid levels occur in major burns which persist up to 3 years post-injury [49, 109–112]. Cytokine levels peak immediately after burn, approaching normal levels only after 1-month post-injury. Constitutive and acute phase proteins are altered beginning 5–7 days post-burn and remain abnormal throughout acute hospital stay. Serum IGF-I, IGFBP-3, parathyroid hormone, and Osteocalcin drop immediately after the injury tenfold, and remain significantly decreased up to 6 months post-burn compared to normal levels [111]. Sex hormones and endogenous growth hormone levels decrease around 3 weeks post-burn [111].
For severely burned patients, the resting metabolic rate at thermal neutral temperature (30 °C) exceeds 140% of normal at admission, reduces to 130% once the wounds are fully healed, then to 120% at 6 months after injury, and 110% at 12 months post-burn [92, 111]. Increases in catabolism result in loss of total body protein, decreased immune defenses, and decreased wound healing [92].
Immediately post-burn patients have low cardiac output characteristic of early shock [113] However, 3–4 days post-burn, cardiac outputs are greater than 1.5 times that of non-burned, healthy volunteers [111]. Heart rates of pediatric burn patients’ approach 1.6 times that of non-burned, healthy volunteers [114]. Post-burn, patients have increased cardiac work [110, 115]. Myocardial oxygen consumption surpasses that of marathon runners and is sustained well into rehabilitative period [115, 116]. There is profound hepatomegaly after injury. The liver increases its size by 225% of normal by 2 weeks post-burn and remains enlarged at discharge by 200% of normal [111].
10% loss of lean body mass is associated with immune dysfunction.
20% loss of lean body mass positively correlates with decreased wound healing.
30% loss of lean body mass leads to increased risk for pneumonia and pressure sores.
40% loss of lean body mass can lead to death [119 #365]
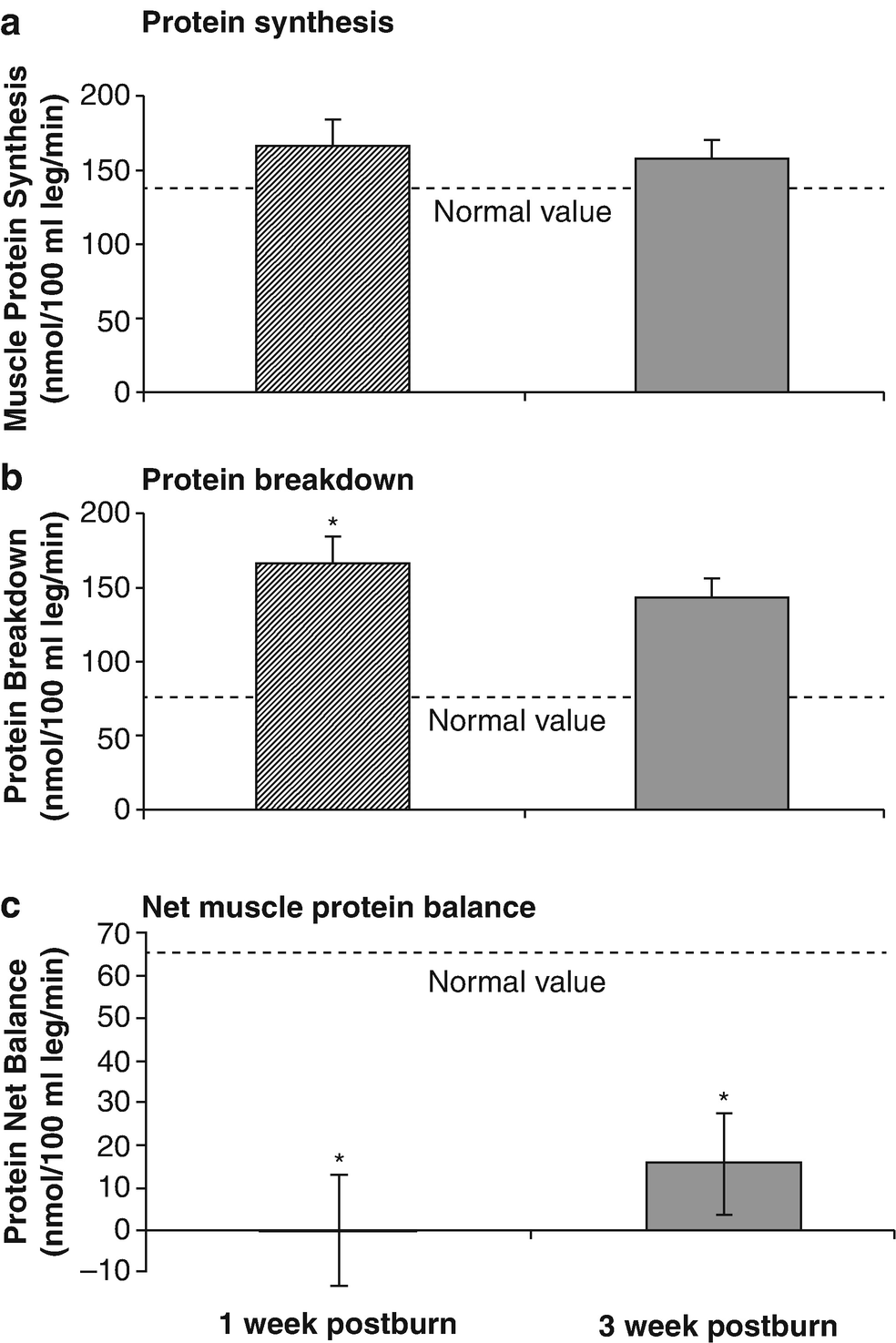
Uncomplicated severely burned patients can lose up to 25% of total body mass after acute burn injury [120]. Protein degradation persists up to nearly 1-year post severe burn injury resulting in significant negative whole body and cross-leg nitrogen balance [110, 118, 121]. Protein catabolism has a positive correlation with increases in metabolic rates [118]. Severely burned patients have a daily nitrogen loss of 20–25 g/ m2 of burned skin [110, 122]. At this rate, a lethal cachexia can be reached in less than 1 month [122]. Burned pediatric patients’ protein loss leads to significant growth retardation for up to 24 months post-injury [123].
Severe burn causes marked changes in body composition during acute hospitalization. Severely burned children lost about 2% of their body weight (−5% LBM, −3% BMC, and −2% BMD) from admission to discharge. Total fat and percent fat increased from admission to discharge by 3% and 7%, respectively.
Septic patients have a particularly profound increase in metabolic rates and protein catabolism up to 40% more compared to those with like-size burns that do not develop sepsis [118]. A vicious cycle develops, as patients that are catabolic are more susceptible to sepsis due to changes in immune function and immune response. Modulation of the hypermetabolic, hypercatabolic response, thus preventing secondary injury is paramount in the restoration of structure and function of severely burned patients.
Elevated circulating levels of catecholamines, glucagon, and cortisol after severe thermal injury stimulate free fatty acids and glycerol from fat, glucose production by the liver, and amino acids from muscle [103, 124, 125]. Specifically, glycolytic-gluconeogenic cycling is increased 250% during the post-burn hypermetabolic response coupled with an increase of 450% in triglyceride-fatty acid cycling [126]. These changes lead to hyperglycemia and impaired insulin sensitivity related to post-receptor insulin resistance demonstrated by elevated levels of insulin, fasting glucose, and significant reductions in glucose clearance [127–130].
18.2.3.3 Glucose Metabolism
Glucose metabolism in healthy subjects is tightly regulated: under normal circumstances, a postprandial increase in blood glucose concentration stimulates release of insulin from pancreatic β-cells. Insulin mediates peripheral glucose uptake into skeletal muscle and adipose tissue and suppresses hepatic gluconeogenesis, thereby maintaining blood glucose homeostasis [131, 132]. In critical illness, however metabolic alterations can cause significant changes in energy substrate metabolism. In order to provide glucose, a major fuel source to vital organs, release of the above-mentioned stress mediators oppose the anabolic actions of insulin [133]. By enhancing adipose tissue lipolysis [125] and skeletal muscle proteolysis [134], they increase gluconeogenic substrates, including glycerol, alanine, and lactate, thus augmenting hepatic glucose production in burned patients [131, 132, 135]. Hyperglycemia fails to suppress hepatic glucose release during this time [136] and the suppressive effect of insulin on hepatic glucose release is attenuated, significantly contributing to post-trauma hyperglycemia [137]. Catecholamine-mediated enhancement of hepatic glycogenolysis, as well as direct sympathetic stimulation of glycogen breakdown, can further aggravate the hyperglycemia in response to stress [132]. Catecholamines have also been shown to impair glucose disposal via alterations of the insulin signaling pathway and GLUT-4 translocation muscle and adipose tissue, resulting in peripheral insulin resistance [131, 138]. Cree and colleagues [137] showed an impaired activation of Insulin Receptor Substrate-1 at its tyrosine binding site and an inhibition of AKT in muscle biopsies of children at 7 days post-burn. Work of Wolfe and colleagues indicates links between impaired liver and muscle mitochondrial oxidative function, altered rates of lipolysis, and impaired insulin signaling post-burn attenuating both the suppressive actions of insulin on hepatic glucose production and on the stimulation of muscle glucose uptake [106, 125, 136, 137]. Another counter-regulatory hormone of interest during stress of the critically ill is glucagon. Glucagon, like epinephrine, leads to increased glucose production through both gluconeogenesis and glycogenolysis [139]. The action of glucagons alone is not maintained over time; however, its action on gluconeogenesis is sustained in an additive manner with the presence of epinephrine, cortisol, and growth hormone [133, 139]. Likewise, epinephrine and glucagon have an additive effect on glycogenolysis [139]. Recent studies found that pro-inflammatory cytokines contribute indirectly to post-burn hyperglycemia via enhancing the release of the above-mentioned stress hormones [140–142]. Other groups showed that inflammatory cytokines, including tumor necrosis factor (TNF), interleukin (IL)-6, and monocyte chemotactic protein (MCP)-1 also act via direct effects on the insulin signal transduction pathway through modification of signaling properties of insulin receptor substrates, contributing to post-burn hyperglycemia via liver and skeletal muscle insulin resistance [143–145]. Alterations in metabolic pathways as well as pro-inflammatory cytokines, such as TNF, have also been implicated in significantly contributing to lean muscle protein breakdown, both during the acute and convalescent phases in response to burn injury [121, 146]. In contrast to starvation, in which lipolysis and ketosis provide energy and protect muscle reserves, burn injury considerably reduces the ability of the body to utilize fat as an energy source.
Skeletal muscle is thus the major source of fuel in the burned patient, which leads to marked wasting of lean body mass (LBM) within days after injury [110, 147]. This muscle breakdown has been demonstrated with whole body and cross-leg nitrogen balance studies in which pronounced negative nitrogen balances persisted for 6 and 9 months after injury [118]. Since skeletal muscle has been shown to be responsible for 70–80% of whole body insulin-stimulated glucose uptake, decreases in muscle mass may significantly contribute to this persistent insulin resistance post-burn [148]. The correlation between hyperglycemia and muscle protein catabolism has been also supported by Flakoll and others [149] in which an isotopic tracer of leucine was utilized to index whole body protein flux in normal volunteers. The group showed a significant increase in proteolysis rates occurring without any alteration in either leucine oxidation or non-oxidative disposal (an estimate of protein synthesis), suggesting an hyperglycemia-induced increase in protein breakdown. Flakoll and others [149] further demonstrated that elevations of plasma glucose levels resulted in a marked stimulation of whole body proteolysis during hyperinsulinemia. A 10–15% loss in lean body mass has been shown to be associated with significant increases in infection rate and marked delays in wound healing [150]. The resultant muscle weakness is further shown to prolong mechanical ventilatory requirements, inhibits sufficient cough reflexes, and delays mobilization in protein-malnourished patients, thus markedly contributing to the incidence of mortality in these patients [151]. Persistent protein catabolism may also account for delay in growth frequently observed in our pediatric patient population for up to 2 years post-burn [123].
Septic patients have a particularly profound increase in metabolic rates and protein catabolism up to 40% more compared to those with like-size burns that do not develop sepsis [92, 152, 153]. A vicious cycle develops, as patients that are catabolic are more susceptible to sepsis due to changes in immune function and immune response. The emergence of multidrug-resistant organisms has led to increases in sepsis, catabolism, and mortality [153–155]. Modulation of the hypermetabolic, hypercatabolic response, thus preventing secondary injury is paramount in the restoration of structure and function of severely burned patients.
Lipid metabolism has recently gained interest in the field of hypermetabolism with the discovery of changes in the structure and function of adipose tissue, called browning. Lipolysis with the change in lipidomic profiling and browning of the adipose could explain in part why hypermetabolism is persistently elevated and is associated with various detrimental outcomes on a clinical level but also on cellular levels.
18.2.3.4 Cardiovascular System: Myocardial Dysfunction
Myocardial function can be compromised after burn injury due to overload of the right heart and direct depression of contractility shown in isolated heart studies [156, 157]. Increases in the afterload of both the left and right heart result from SVR and PVR elevations. The left ventricle compensates and CO can be maintained with increased afterload by augmented adrenergic stimulation and increased myocardial oxygen extraction. The right ventricle has a minimal capacity to compensate for increased afterload. In severe cases, desynchronization of the right and left ventricles is deleteriously superimposed on a depressed myocardium [158]. Burn injury greater than 45% TBSA can produce intrinsic contractile defects. Several investigators reported that aggressive early and sustained fluid resuscitation failed to correct left ventricular contractile and compliance defects [157–159]. These data suggest that hypovolemia is not the only mechanism underlying the myocardial defects observed with burn shock. Serum from patients failing to sustain a normal CO after thermal injury have exhibited a markedly negative inotropic effect on in vitro heart preparations, which is likely due to the previously described shock factor [160]. In other patients with large burn injuries and normal cardiac indices, little or no depressant activity was detected.
Sugi and colleagues studied intact, chronically instrumented sheep after a 40% TBSA flame burn injury and smoke inhalation injury, and smoke inhalation injury alone. They found that maximal contractile effects were reduced after either burn injury or inhalation injury [161, 162]. Horton and others demonstrated decreased left ventricular contractility in isolated, coronary perfused, guinea pig hearts harvested 24 h after burn injury [163]. This dysfunction was more pronounced in hearts from aged animals and was not reversed by resuscitation with isotonic fluid. It was largely reversed by treatment with 4 mL/kg of hypertonic saline dextran (HSD), but only if administered during the initial 4–6 h of resuscitation [164, 165]. These authors also effectively ameliorated the cardiac dysfunction of thermal injury with infusions of antioxidants, arginine and calcium channel blockers [166–168]. Cioffi and colleagues in a similar model observed persistent myocardial depression after burn when the animals received no resuscitation after burn injury [169]. As opposed to most studies, Cioffi reported that immediate and full resuscitation totally reversed abnormalities of contraction and relaxation after burn injury. Murphy et al. showed elevations of a serum marker for cardiac injury, Troponin I, for patients with a TBSA >18%, despite good cardiac indices [170]. Resuscitation and cardiac function studies emphasize the importance of early and adequate fluid therapy and suggest that functional myocardial depression after burn injury may not occur in patients receiving prompt and adequate volume therapy.
The primary mechanisms by which burn shock alters myocardial cell membrane integrity and impairs mechanical function remain unclear. Oxygen-derived free radicals may play a key causative role in the cell membrane dysfunction that is characteristic of several low-flow states. Horton et al. showed that a combination therapy of free radical scavengers SOD and catalase significantly improved burn-mediated defects in left ventricular contractility and relaxation when administered along with adequate fluid resuscitation (4 mL/kg per percent of burn). Antioxidant therapy did not alter the volume of fluid resuscitation required after burn injury [166].
18.2.3.5 Effects on the Renal System
Diminished blood volume and cardiac output result in decreased renal blood flow and glomerular filtration rate. Other stress-induced hormones and mediators such as angiotensin, aldosterone, and vasopressin further reduce renal blood flow immediately after the injury. These effects result in oliguria, which, if left untreated will cause acute tubular necrosis and renal failure. Twenty years ago, acute renal failure in burn injuries was almost always fatal. Today newer techniques in dialysis became widely used to support the kidneys during recovery [171]. The latest reports indicate an 88% mortality rate for severely burned adults and a 56% mortality rate for severely burned children in whom renal failure develops in the post-burn period [172, 173]. Early resuscitation decreases risks of renal failure and improves the associated morbidity and mortality [174].
18.2.3.6 Effects on the Gastrointestinal System
The gastrointestinal response to burn is highlighted by mucosal atrophy, changes in digestive absorption, and increased intestinal permeability [175]. Atrophy of the small bowel mucosa occurs within 12 h of injury in proportion to the burn size and is related to increased epithelial cell death by apoptosis [176]. The cytoskeleton of the mucosal brush border undergoes atrophic changes associated with vesiculation of microvilli and disruption of the terminal web actin filaments. These findings were most pronounced 18 h after injury, which suggests that changes in the cytoskeleton, such as those associated with cell death by apoptosis, are processes involved in the changed gut mucosa [177]. Burn also causes reduced uptake of glucose and amino acids, decreased absorption of fatty acids, and reduction in brush border lipase activity [178]. These changes peak in the first several hours after burn and return to normal at 48–72 h after injury, a timing that parallels mucosal atrophy.
Intestinal permeability to macromolecules, which are normally repelled by an intact mucosal barrier, increases after burn [179]. Intestinal permeability to polyethylene glycol 3350, lactulose, and mannitol increases after injury, correlating to the extent of the burn [180]. Gut permeability increases even further when burn wounds become infected. A study using fluorescent dextrans showed that larger molecules appeared to cross the mucosa between the cells, whereas the smaller molecules traversed the mucosa through the epithelial cells, presumably by pinocytosis and vesiculation [181]. Mucosal permeability also paralleled increases in gut epithelial apoptosis.
Changes in gut blood flow are related to changes in permeability. Intestinal blood flow was shown to decrease in animals, a change that was associated with increased gut permeability at 5 h after burn [182]. This effect was abolished at 24 h. Systolic hypotension has been shown to occur in the hours immediately after burn in animals with a 40% TBSA full-thickness injury. These animals showed an inverse correlation between blood flow and permeability to intact Candida [183].
The best treatment to alleviate mucosal atrophy is early initiation of enteral nutrition, usually within 8–12 h post-burn. Glutamine and other antioxidants have been shown to improve enteral inflammatory driven pathways as well as gut function.
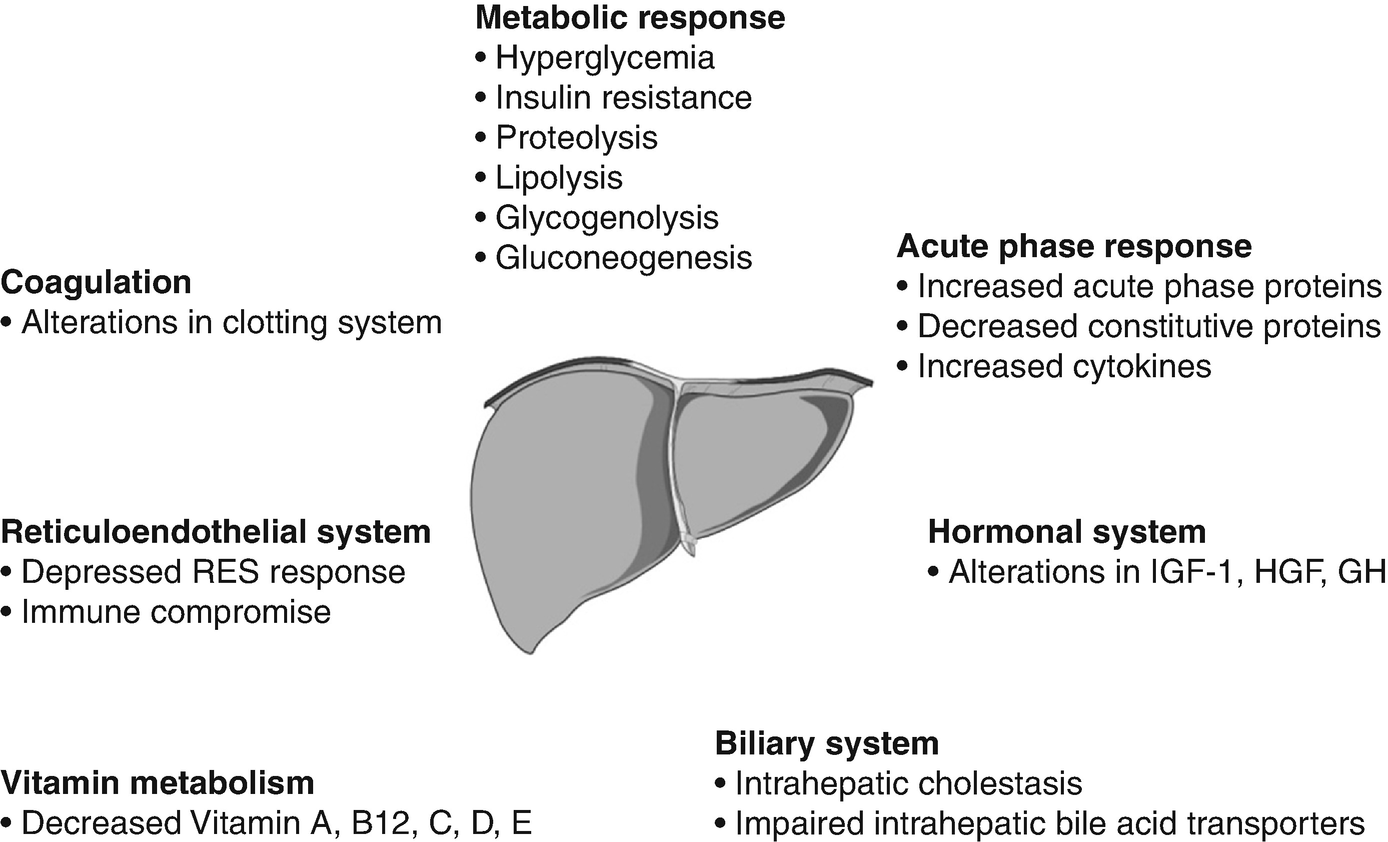
Despite the need for liver function and integrity the liver is profoundly affected post-burn and in our opinion a central contributor to post-burn morbidity and mortality [111, 184]. The liver has several myriad functions that are each essential for survival:
Related posts:
 of Burn Patients to the Burn Center Including Burn Wound Evaluation
of Burn Patients to the Burn Center Including Burn Wound Evaluation
 Care of the Burned Patient
Care of the Burned Patient
 Management of Facial Burns, Acute Versus Long-Term, Surgical Versus Non-surgical Face Transplant
Management of Facial Burns, Acute Versus Long-Term, Surgical Versus Non-surgical Face Transplant
 Management of Burn Patients and Fluid Resuscitation
Management of Burn Patients and Fluid Resuscitation
 Improvement in Burn Care
Improvement in Burn Care
 Necrolysis Spectrum from Basic Theory to Practice Essentials
Necrolysis Spectrum from Basic Theory to Practice Essentials
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


