Fig. 45.1
Symblepharon in a patient with OcMMP
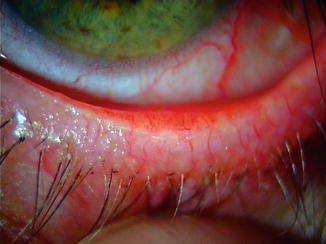
Fig. 45.2
Blepharitis in the lower eye lid margin
The course of OcMMP is variable among patients. Some patients have limited disease and mild ocular scarring with remission following treatment, while approximately one-third of patients with OcMMP experience chronic, progressive disease with only partial response to treatment, necessitating lifelong follow-up [7, 16, 17]. The disease is also more progressive in younger patients. Rauz et al. examined the clinical features of 18 young patients (under 60 years of age) with OcMMP and compared the findings with 18 patients of ages 70 years and above. The authors demonstrated that mucocutaneous involvement was more common in the younger age group, with more severe ocular staging at presentation. The latter patients also required immunosuppressive therapy earlier than the older patient group, and despite this, their disease progressed more rapidly [18].
It is imperative to quantify the extent of fibrosis and conjunctival inflammation in the disease, to assess progression of the fibrotic process and efficacy of treatment. Two established staging systems are currently in use to quantify the degree of fibrosis and conjunctival inflammation in ocular MMP—the Mondino [19] and Foster [6] system. The Mondino system uses the degree of lower fornix shortening, while the Foster system is based on the presence of specific clinical signs. Tauber et al. cited the relative insensitivity of both systems in detecting disease progression and proposed a new system that encompassed elements from both the Mondino and the Foster systems. This includes counting the number of symblephara and the percentage horizontal obliteration of the lower fornix [20] (Tables 45.1 and 45.2).
System | Characteristics |
|---|---|
Foster stages | I. Subconjunctival scarring and fibrosis |
II. Fornix foreshortening of any degree | |
III. Presence of symblepharon, any degree | |
IV. Ankyloblepharon, frozen globe | |
Mondino stages | I. 0–25 % loss of inferior fornix depth |
II. 25–50 % loss of inferior fornix depth | |
III. 50–75 % loss of inferior fornix depth | |
IV. 75–100 % loss of inferior fornix depth |
Table 45.2
Proposed staging system by Tauber et al.
System | Characteristics | |
|---|---|---|
I | Subconjunctival scarring and fibrosis | |
II | a | 0–25 % loss of inferior fornix depth |
b | 25–50 % loss of inferior fornix depth | |
c | 50–75 % loss of inferior fornix depth | |
d | 75–100 % loss of inferior fornix depth | |
III | a | 0–25 % horizontal involvement of symblephara |
b | 25–50 % horizontal involvement of symblephara | |
c | 50–75 % horizontal involvement of symblephara | |
d | 75–100 % horizontal involvement of symblephara | |
n | Number of symblephara countable | |
IV | Ankyloblepharon, frozen globe |
45.3 Linear Immunoglobulin A Bullous Dermatosis (LABD)
LABD is a rare subepidermal blistering disease characterised by subepidermal blister formation and linear deposition of immunoglobulin A basement membrane antibodies. The target antigen is a key component of the dermoepidermal adhesion complex [21]. The mucous membranes including the eye and mouth are involved in approximately 80 % of cases [22]. Ocular involvement is estimated to occur in around 50 % of patients [23]. Both eyes are usually involved, although it may be asymmetrical. Ocular symptoms include eye pain, redness, itching, burning, dry eye, foreign body sensation and mucus discharge. Ocular findings may be clinically indistinguishable from those in OcMMP. Slit lamp examination may reveal conjunctival subepithelial fibrosis and subsequent loss of inner canthal architecture, fornix foreshortening and symblepharon formation. Other findings include conjunctival bullae, entropion, trichiasis, corneal opacification and blindness [24, 25].
45.4 Epidermolysis Bullosa Acquisita (EBA)
EBA is a very rare subepidermal blistering disorder characterised by IgG autoantibodies to type VII collagen, a primary component of the BMZ anchoring fibril [26–29]. Circulating antibodies are seen in 20–50 % of patients with EBA, and IgA antibodies are seen even less frequently [30]. In some patients with EBA, IgA antibodies are more abundant than those of the IgG class or are the only immunoglobulin class detected—this is termed “IgA-EBA” [31–33].
Mucosal involvement in EBA is common and primarily oral [22]. Ocular manifestations include bilateral subepithelial vesicles, conjunctival erosion, conjunctival scarring, blepharitis and cicatricial trichiasis. Corneal abnormalities include peripheral ulcerative keratitis, corneal neovascularisation, corneal thinning, corneal perforation, corneal scarring, corneal pannus (Fig. 45.3) and opacification [22, 24, 34]. The IgA-EBA form of the disease is associated with more severe ocular involvement [31–33], with total blindness reported to occur in these patients [35, 36].
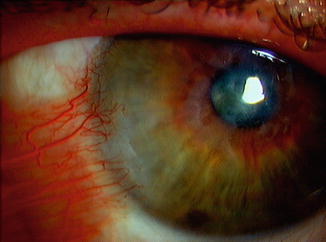
Fig. 45.3
Cornea pannus
45.5 Other Autoimmune Blistering Diseases
Ocular complications are seen less commonly in the other types of autoimmune blistering diseases.
In pemphigus vulgaris, the mucous membranes are usually the first affected by the disease. Erosions in the oral cavity occur commonly. The mucosal surfaces including the conjunctiva, oesophagus and genital areas can also be affected, but are less common. Ocular involvement in pemphigus is rare [11]. It may however precede disease manifestations at other sites [37]. Ocular disease can present with symptoms of ocular irritation, tearing and foreign body sensation [37–39]. Ocular findings reported include conjunctivitis with mucoid discharge, conjunctival oedema and conjunctival vesicles [40]. In a case series of 11 patients with PV, Daoud et al. reported ocular findings of bilateral conjunctivitis and lid margin ulceration. The mean interval between onset of PV and ocular involvement was 20 months, and the ocular disease lasted for a mean of 12 months. The authors found that patients had complete recovery and no long-term sequelae due to conjunctival disease. Based on their cohort of 167 patients with PV, Daoud et al. estimated an incidence of ocular involvement in pemphigus vulgaris of 7 %. Fornix foreshortening and plica semilunaris vegetations have also been reported by other authors in the literature [41]. Ocular disease does not appear to affect visual acuity, and patients usually have a full recovery without long-term complications [42]. Pemphigus foliaceus, a separate type of pemphigus, may produce entropion and trichiasis of both eyelids with resultant corneal damage (Fig. 45.4). Progressive scarring and blindness usually do not occur [43].
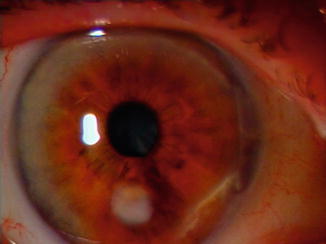
Fig. 45.4
Inferior corneal scar in a patient with Pemphigus Vulgaris
Mucous membranes are involved only occasionally in bullous pemphigoid; consequently, ocular involvement is uncommon. Ocular manifestations are subtle and may go undetected in the conjunctiva and lid margins. Slit lamp examination may reveal subepithelial fibrosis with fine linear striae of the tarsus [44, 45]. Other ocular manifestations reported in the literature include conjunctival scarring, corneal epithelial defects, stroma oedema and corneal opacity [44, 45]. Corneal melt [46] and filamentary keratitis [47] have been reported in paraneoplastic pemphigus. In dermatitis herpetiformis, ophthalmic involvement is rare and is limited to the periorbital skin [48].
45.6 Treatment
Treatment in autoimmune blistering skin diseases is individualised based on age, disease severity and the sites of involvement in the patient. In patients who present with early-stage disease, topical corticosteroids such as triamcinolone acetonide, fluocinonide or clobetasol propionate may be used. Topical corticosteroids are however inadequate to halt disease progression. Systemic chemotherapy is used for acute exacerbations and in chronic disease. Dapsone is used in milder cases unless the patient has a history of glucose-6-phosphate dehydrogenase deficiency or drug intolerance (Refs. [35, 36] in Elchahal). In moderate to severe cases, cytotoxic agents are used to suppress conjunctival inflammation and cicatrisation. These agents include mycophenolate mofetil, sulphasalazine, methotrexate and azathioprine. Cyclophosphamide together with prednisolone at a dose of 1 mg per 1 kg of body weight per day is used in resistant cases. In patients whose disease is refractory to immunosuppressive therapy, intravenous immunoglobulin (IVIG) can be used. It is administered at a dose of 2–3 g per 1 kg of body weight per cycle, with each cycle lasting for three to five consecutive days each month. Adverse events associated with IVIG therapy are usually mild and self-limiting. The use of IVIG therapy is however limited due to its high cost. If the cost of management of side effects and hospitalisations are included however, IVIG has been shown to be more cost-effective than immunosuppressive therapy over the entire disease course and an on annual basis. Etanercept and infliximab are other treatment options in disease refractory to corticosteroids or immunosuppressive drugs. Surgical techniques are used in cases of MMP with advanced corneal keratinisation or ulceration. These include amniotic membrane grafting, penetrating keratoplasty and limbal stem cell transplantation. If these techniques are unsuccessful, keratoprostheses may be another option to regain useful vision in the affected eye.
45.7 Conclusion
Ocular involvement in AIBD varies in frequency depending on the type of AIBD, with mucous membrane pemphigoid being the most frequent type. The clinical features in the eye overlap between these types and may mimic other milder non-scarring inflammatory ocular diseases. Early recognition and treatment to prevent irreversible scarring are essential.
Related posts:
 Kindlin-1 and Its Role in Kindler Syndrome
Kindlin-1 and Its Role in Kindler Syndrome
 Cyclophosphamide in Autoimmune Blistering Diseases: Safety, Efficacy and Evidence Base
Management of Bullous Systemic Lupus Erythematosus
Cyclophosphamide in Autoimmune Blistering Diseases: Safety, Efficacy and Evidence Base
Management of Bullous Systemic Lupus Erythematosus
 Using Intravenous Immunoglobulins in Autoimmune Bullous Diseases
Using Intravenous Immunoglobulins in Autoimmune Bullous Diseases
 Living with Epidermolysis Bullosa: Reviewing the Impact on Individuals’ Quality of Life
Living with Epidermolysis Bullosa: Reviewing the Impact on Individuals’ Quality of Life
 How to Take a Skin Biopsy Correctly to Diagnose Epidermolysis Bullosa and Autoimmune Bullous Diseases
How to Take a Skin Biopsy Correctly to Diagnose Epidermolysis Bullosa and Autoimmune Bullous Diseases
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


