34 Nonsyndromic craniosynostosis
Synopsis
 Craniosynostosis is the pathologic fusion of one or more cranial vault sutures, usually resulting in an abnormal head shape. Nonsyndromic craniosynostosis occurs in a sporadic, nonfamilial fashion, in the absence of an associated genetic syndrome.
Craniosynostosis is the pathologic fusion of one or more cranial vault sutures, usually resulting in an abnormal head shape. Nonsyndromic craniosynostosis occurs in a sporadic, nonfamilial fashion, in the absence of an associated genetic syndrome.





 Complications can be divided into early or late events.
Complications can be divided into early or late events.
 Secondary revisions may be necessary involving either the soft tissue, bone, or both.
Secondary revisions may be necessary involving either the soft tissue, bone, or both.

Introduction
Craniosynostosis is the pathologic fusion of one or more cranial vault sutures, usually resulting in an abnormal head shape. Nonsyndromic craniosynostosis refers to an isolated entity, without an associated genetic syndrome, occurring in approximately 1 in 1800 to 1 in 2500 births. Typically in nonsyndromic forms only one suture is involved, termed simple craniosynostosis, though occasionally two or more sutures may be affected (complex craniosynostosis). The type of synostosis is denoted based on the suture fused, with sagittal being the most common and lambdoid the least. There is recent evidence that the distribution of metopic and unicoronal synostosis (UCS) is changing, with a relative increase in metopic craniosynostosis.1,2 Midline synostoses (sagittal and metopic) appear more apt to occur among twins.3 Gender predilection demonstrates male preference for sagittal synostosis (4 : 1), while females are slightly more prone to unilateral coronal synostosis (3 : 2).4 A complex, multifactorial set of etiologic factors is at work in nonsyndromic craniosynostosis given the differences in population incidence, proportion of family cases, male-to-female distribution, and phenotypic severity among and between types of sutural synostosis.5
Historical Perspective
Craniosynostosis has been in existence since antiquity. Evidence of fused cranial sutures is documented in skull specimens from both prehistoric and near-historic times, representing multiple regions and populations.6–10 Likely, the disease mechanism has been conserved through evolutionary history. However, cultural practices, including artificial head molding, may have contributed to increased prevalence in certain population groups.11
Prominent historical figures are posited to have been afflicted by craniosynostosis. Akhenaten, the renowned Egyptian pharaoh, is described and depicted as having an elongated head shape with an overhanging occiput. His dolichocephalic cranial form is thought to represent sagittal synostosis, and is shared by several people in his genetic lineage (e.g., Tutankhamun and Thutmose III).12 The Greek politican Pericles is also considered to have had sagittal synostosis.13 Artistic renderings of Pericles all show him with a helmet pulled up above his eyes, hiding his head shape. Close inspection reveals presence of bitemporal narrowing and possibly hypotelorism. The ancient Greek biographer Plutarch described Pericles as: “overall handsome but with the head enormously long … for this reason, all the statues representing him wear a helmet because the artists did not want to put into evidence such a defect.”14
The first description of a fused cranial suture was by Otto in 1830.15 It was in this report that the term “craniosynostosis” was initially penned. Recognizing that early fusion of a cranial suture caused development of an abnormal head shape was put forth by Stahl and Hyrtl.16 Virchow was the first to classify the different patterns of skull deformity and introduced the morphologic terms still in use today.16,17 Virchow also postulated on the underlying cause of craniosynostosis, and examined cranial growth mechanisms.16,17 Marie Lannelongue was the first to report on a surgical technique to manage craniosynostosis, advocating to release (not resect) the fused suture.18 The strip craniectomy was first described a short time later; however, the initial case was complicated by mortality.19 Mehner championed the first successful use of the strip craniectomy to remove the fused suture.20 This technique was expounded upon by Faber and Towne, who performed more radical craniectomy techniques.21 Shillito and Matson contributed an early large series on surgically treated patients with craniosynostosis.22 Tessier23,24 and Rougerie et al.25 furthered efforts by incorporating aesthetic objectives into the treatment of craniosynostosis.
Basic science/disease process
Nonsyndromic craniosynostosis most frequently occurs in a sporadic and nonfamilial fashion. The mechanistic cause and biochemical changes at the suture appear to stem from a variety of genetic and environmental factors. The genetic influence, however, is poorly understood. Ephrin-A4 (EFNA4) is, as yet, the only identified gene proposed to play a role in nonsyndromic craniosynostosis.26 Autosomal-dominant familial inheritance, in the absence of a known identifiable gene, is reported to account for approximately 8–14% of nonsyndromic synostoses.5,27
About 2% of sagittal synostosis cases are familial.28 Coronal synostosis demonstrates an 8–10% positive family history.5 Bicoronal synostosis is more apt to be inherited than UCS.27 Older paternal age may contribute more to coronal than sagittal synostosis. Metopic synostosis is thought to be familial in up to 10% of cases.29,30 Seemingly nonsyndromic coronal and metopic synostosis can be associated with a subtle presentation of phenotypic features in Muenke and Saethre–Chotzen syndromes. FGFR3 and TWIST gene mutations should be tested in cases of craniosynostosis with suspected familial inheritance.26,31 Lambdoid synostosis occurs so infrequently that it is difficult to quantify the incidence of heritable cases. There have been a few case reports denoting lambdoidal synostosis within families.32–34
Environmental effects on suture biology are also suggested to promote synostosis. Antenatal head compression, from multiple gestation, large infant size, abnormal intrauterine lie, or uterine abnormalities, has been reported in association with nonsyndromic craniosynostosis. Graham and Smith describe two cases of metopic synostosis and one case of coronal synostosis secondary to bicornuate uterine morphology, cephalic compression in the pelvis from a triplet gestation, and a constricted in utero lie after early descent, respectively.35 Higginbottom et al. recounted three cases of craniosynostosis also resulting from external force to the head, one each from breech position, amniotic band, and a morphologic abnormality of the uterus.36 Twin studies demonstrate an increase in craniosynostosis, providing further evidence that gestational constraint contributes to premature sutural fusion in all twin types, while monozygotic twins may have the additional influence of genetic factors.5
The concept of intrauterine constraint is borne out in experimental models. Animal studies demonstrate that intrauterine constraint results in 88% suture fusion, with FGFR2 and transforming growth factor-β (TGF-β) expression being enhanced in the fused sutures.37–39 In addition to TGF-β, other growth factors shown to upregulate from head constraint, possibly influencing sutural stenosis, include: BMP-4, Noggin, and Indian hedgehog.38,40,41 However, restriction of sutural expansion in lambs by rigid plating across the coronal sutures 8 weeks antepartum demonstrated persistently patent sutures, by CT scan and histologically, despite the phenotypic appearance of bicoronal synostosis.42
A host of other nongenetic risk factors have been reported in association with nonsyndromic craniosynostosis. Maternal smoking, white maternal race, advanced maternal age, gestation at high altitude, use of nitrosatable drugs (e.g., nitrofurantoin), paternal occupation (e.g., agriculture, forestry), fertility treatments, endocrine abnormalities (e.g., hyperthyroidism), and warfarin ingestion during gestation have all been linked to craniosynostosis.43–48
Regardless of the genetic and environmental influences, it is theorized that fusion of the suture occurs as either a primary or secondary event. Ossification of the suture as the principal cause was first suggested by Virchow, and others have since agreed.16,17,49–52 An increased number of osteoblasts at the affected suture results in calvarial ossification and subsequent suture fusion.49,50,52–55 This concept intimates that any change in cranial base length, brain volume, cerebrospinal fluid (CSF) volume, and ICP are secondary to the primary event at the suture (sutural fusion).
The fusion of the suture as a secondary issue to either cranial base underdevelopment or diminished brain growth has also been proposed. Gunther, in 1931, first suggested that delayed growth of the cranial base was the first step leading to sutural fusion.56 Moss furthered this theory, stating that the skull base abnormality promoted tight dural attachments adjacent to the vault sutures.57 He postulated that suture fusion results when these dural connections tether the osseous plates and prevent the stretch at the cranial sutures prompted by normal brain growth.58,59 This implies that diminished cranial base length is the primary problem resulting in secondary sutural fusion, and possible changes to the brain and CSF volume.
Lack of intrinsic brain growth potential has also been suggested to cause early fusion of cranial sutures.59 Diminished brain growth subjacent to the suture, hence less stimulus and stretch on the vault sutures, may portend sutural fusion. Marsh et al. compared a series of infants with nonsyndromic coronal synostosis to normal control infants on CT scan, demonstrating dysmorphic brain features in the former group.60
Fellows-Mayle and colleagues tested the three theories of sutural fusion in a rabbit model using the Path analysis.61 The primary suture fusion model was found to describe best the causal nature of both early- and delayed-onset craniosynostosis. In any event, once the suture ossifies early, a downstream series of events is initiated with aesthetic, growth-related, and functional consequences.
The events that succeed the fused cranial suture relate to restriction of brain expansion in the area subjacent to the stenosis. Skull expansion is restricted in vectors perpendicular to the fused suture and, with forces directed elsewhere, compensatory bulges develop, usually parallel to the fused suture.16,17 Delshaw et al. outlined four concepts describing the changes in cranial vault expansion that occur as a result of diminished growth at the fused suture.62,63
1. “Cranial vault bones that are prematurely fused act as a single bone plate with decreased growth potential.
2. Abnormal asymmetrical bone deposition occurs at perimeter sutures with increased bone deposition directed away from the bone plate.
3. Perimeter sutures adjacent to prematurely fused suture compensate in growth more than perimeter sutures distant to sutural stenosis.
4. A nonperimeter suture that is contiguous to the premature fused suture undergoes enhanced symmetrical bone deposition along both edges.”
More significantly, the fusion of cranial sutures is associated with increased ICP. The risk with a single suture synostosis is less than with multiple sutures (14% versus 47%), but a valid risk nonetheless in all cases.64 Recent reports demonstrate that, even in mild cases of nonsyndromic synostosis, a significant proportion exhibit intracranial hypertension (defined as >17 mmHg) on lumbar puncture.65 Increased ICP occurs because the compartment (cranial vault) is too tight for its contents (brain). Additionally, there may be a discrepancy between CSF production and egress. The pressure on the brain portends neurologic sequelae, including headaches, nausea, vomiting, visual problems, and motor, behavioral, and intellectual delay.
Diagnosis/patient presentation
Physical examination is the cornerstone in the diagnosis of craniosynostosis. Symmetric brain enlargement is inhibited by the fused sutures and compensatory cranial bulges occur where sutures are patent. The cranium takes on a characteristic shape depending on the suture(s) involved (Fig. 34.1). Palpation and sometimes even visual inspection can reveal the presence of ridging at fused suture lines. Head circumference and cephalic index are adjunctive measurements that may support the diagnosis (Table 34.1). However, visual inspection and palpation remain the most effective means of confirming the diagnosis. Fusion of multiple sutures should raise greater concern for a genetic or familial cause and for intracranial hypertension.
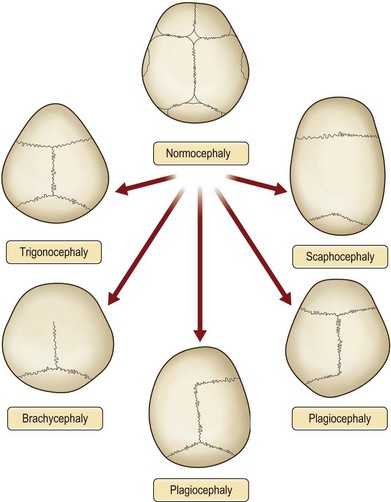
Table 34.1 Cephalic index (CI), based on maximal cranial length and maximal cranial width: CI = Wd/L × 100, where Wd is the maximal cranial width and L is the maximal cranial length
| Cephalic index | Suggested diagnosis |
|---|---|
| <71 | Hyperdolichocephalic |
| 71–76 | Dolichocephalic/scaphocephalic (as with sagittal) |
| 76–81 | Mesocephalic |
| 81–81.5 | Brachycephalic (as with bicoronal) |
| >85.5 | Hyperbrachycephalic |
(Reproduced from Bennaceur S, Petavy-Blanc AS, Chauve J, et al. Human cephalic morphology. Anthropometry. In: Laffont A, Durieuz F, eds, Encyclopédie médicochirurgicale. Elsevier; 2005:85–103).
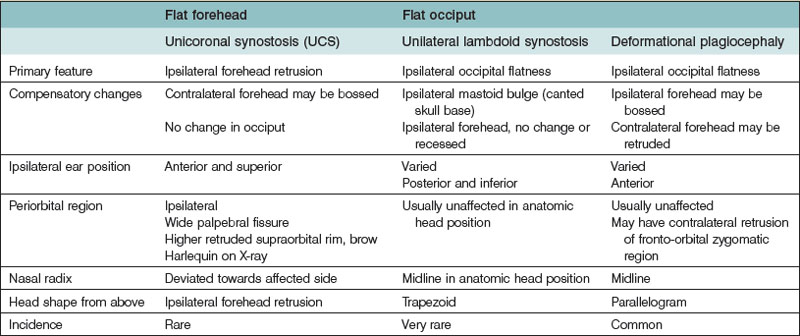
True craniosynostosis must be differentiated from positional plagiocephaly. A comprehensive discussion of deformational plagiocephaly is outside the scope of this chapter, but we will address important distinguishing features from synostotic plagiocephaly (Table 34.2). Deformational occipital flatness has risen dramatically following the “back to sleep” campaign advocated by the American Academy of Pediatrics.66 Infants with positional plagiocephaly usually have a history of sleeping supine with the head consistently turned toward one side.67 Torticollis frequently occurs with positional plagiocephaly. The shortened sternocleidomastoid muscle favors ipsilateral head turning, or conversely, the ipsilaterally flattened occiput leads to the torticollis secondarily.68
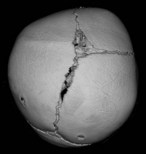
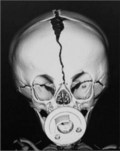
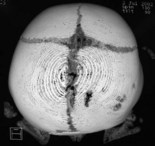
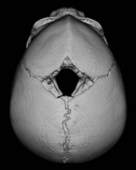
When clinical suspicion strongly supports the diagnosis, a three-dimensional (3D) CT scan is a necessary adjunct to document the presence and extent of synostosis. A high-resolution scan will readily illustrate the fused suture and allow analysis from several angles. The CT scan also allows for assessment of ventricular size, defects of the corpus callosum, and presence of brainstem abnormalities. Importantly, radiologic signs of increased ICP can be witnessed. The characteristic “thumbprinting” or “copper-beaten” patterns, when evident on 3D scan or the scout film, suggest raised cranial pressure (Fig. 34.2). Similarly, 2D cuts can show loss of gyral folding and blunted cisternae along the endocranial surface, consistent with the brain trying to expand against an immobile, restrictive cranial vault. The CT scan also may aid in surgical planning and serves as a baseline study to compare against postoperative changes.
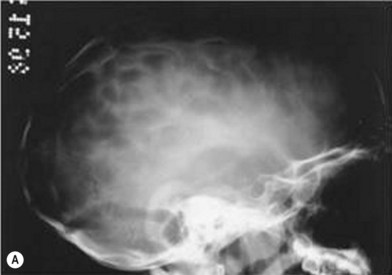
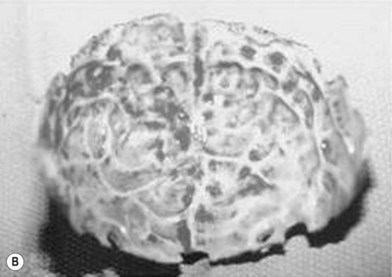
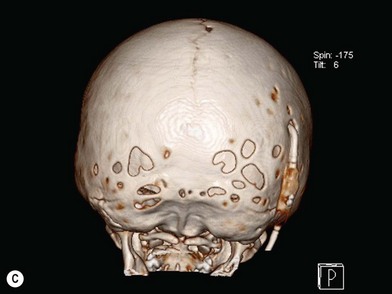
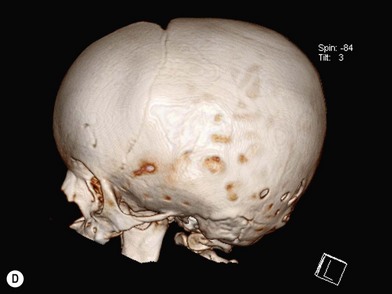
Fig. 34.2 Radiologic signs of increased cranial pressure – “thumbprinting.”
(Reproduced from Weinzweig J, Baker SB, Whitaker LA, et al. Delayed cranial vault reconstruction for sagittal synostosis in older children: an algorithm for tailoring the reconstructive approach to the craniofacial deformity. Plast Reconstr Surg. 2002;110:397–408.)
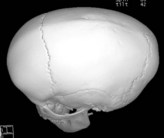
Sagittal synostosis develops following fusion of the midline, sagittal suture (Table 34.3). This results in a “boat-shaped” head (i.e., scaphocephaly) with an expanded anteroposterior dimension and a narrowed bitemporal distance. Cephalic index is consistent with dolichocephaly (<76). A sagittal ridge may be appreciated on palpation. Fusion of the anterior sagittal suture contributes to significant frontal bossing. Similarly, an occipital bulge results from involvement of the posterior sagittal suture. Varying degrees of frontal or occipital prominence can occur depending on the extent and location of synostosis along the sagittal suture.
| Suture fused | Features |
|---|---|
| Sagittal | Elongated anteroposterior head |
| Frontal and occipital prominence | |
| Biparietal narrowness | |
 |  |
 |  |
Plagiocephaly is a general term denoting an asymmetric coronal plane of the cranium. The twisted head shape is the result of unilateral coronal synostosis, unilateral lambdoid synostosis, or positional molding (Table 34.2). Some object to the term “plagiocephaly” as anatomically imprecise. Referral by the affected suture is the preferred terminology, but the general phenotypic descriptor “plagiocephaly” is still often encountered.
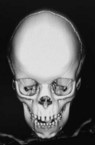
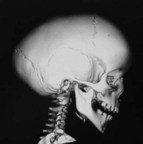
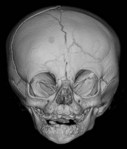
UCS is termed “anterior plagiocephaly” (Table 34.4). UCS demonstrates a flattened ipsilateral forehead and supraorbital rim, a raised eyebrow, and a widened palpebral opening on the affected side. The lateral orbital rim and ipsilateral temporal area also appear deficient. The root of the nose deviates toward the affected suture. The ear may be displaced anterior and superior on the side of coronal fusion. The contralateral forehead is often bossed, which is a compensatory change. The occipital region is usually unaffected and symmetric, which, along with the pathognomonic orbital changes, differentiates UCS from other causes of plagiocephaly. Facial asymmetry can occur, particularly if the UCS is left unchecked. When present, the chin deviates to the unaffected side secondary to changed position of the glenoid fossa.63,69 The affected orbit is vertically taller and narrower than the contralateral side on frontal 3D CT. Intraorbitally, the ipsilateral steep superior orbital fissure and sphenoid wing is known as the harlequin deformity. Patency of the frontosphenoidal suture should also be investigated on CT scan as the disease process can continue to this terminal extension of the coronal suture. In rare cases, frontosphenoidal synostosis alone can result in morphology that mimics UCS.70
Table 34.4 Anterior plagiocephaly
| Suture fused | Features |
|---|---|
| Unilateral coronal | Ipsilateral |
| Retruded forehead and supraorbital rim | |
| Raised eyebrow | |
| Widened palpebral fissure | |
| Radix deviates to affected ear anterior and superior | |
| Contralateral forehead bossed | |
 |  |
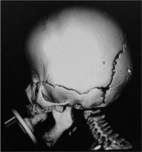
Bicoronal synostosis is symmetric, does not cause plagiocephaly, and is more likely to be either familial nonsyndromic or associated with a syndrome (Table 34.5). Fusion of both coronal sutures results in brachycephaly (flat head), marked by increased biparietal diameter, and decreased anteroposterior diameter leading to a blunted forehead and supraorbital ridge. In certain cases, there can be a compensatory increase in parietal height causing turribrachycephaly (tall flat head).
| Suture fused | Features |
|---|---|
| Bilateral coronal | Bilateral retruded forehead and supraorbital rims |
| Increased biparietal diameter | |
 |  |
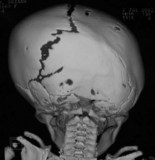
Unilateral lambdoidal synostosis is a very rare cause of plagiocephaly (“posterior plagiocephaly”) (Table 34.6). It is characterized by a cant of the posterior skull base with occipital flatness on the affected side and an ipsilateral inferiorly displaced mastoid bulge. In some cases there is secondary contralateral forehead prominence, providing an overall trapezoid head shape when viewed from above.71 Clinically, the ipsilateral ear position is inferior, but quite varied in terms of anteroposterior placement. However, CT scan always shows the affected ear is closer to the anterior nasal spine.72–74 CT also demonstrates deviation of the posterior cranial fossa toward the affected lambdoid suture, with a symmetric anterior fossa.
Table 34.6 Posterior plagiocephaly
| Suture fused | Features |
|---|---|
| Unilateral lambdoid | Ipsilateral occipital flatness |
| Ipsilateral mastoid bulge | |
| Ear inferior | |
 |  |
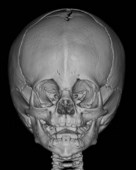
Trigonocephaly refers to the triangular-shaped head that occurs with early fusion of the metopic suture (Table 34.7). The metopic suture ordinarily closes at 8 months of life.75 The spectrum of severity can range from a metopic ridge alone to significant ridging with marked trigonocephaly. The features of metopic synostosis include a keel-shaped forehead, bitemporal narrowing, parietal expansion, supraorbital and lateral orbital retrusion, and hypotelorism. Metopic synostosis is most apt to be associated with midline brain aberrations and one report suggests a higher than usual concomitant presentation of Chiari I malformation.76
| Suture fused | Features |
|---|---|
| Metopic | Keel-shaped forehead |
| Bitemporal narrowing | |
| Hypotelorism | |
 |  |
Patient selection
The principal functional concerns relate to ICP elevation which can lead to neurologic impairment (e.g., developmental delay, visual loss). Treatment of the synostotic cranium is intended to avoid such potential brain malfunction related to increased ICP.77 This raises the question of the utility of preoperative ICP monitoring. In cases where the parents are committed to surgical correction of the child and/or in the presence of papilledema or other suggestive symptoms or signs of intracranial hypertension, subjecting the infant to formal ICP monitoring is not warranted. However, when parents are uncertain if they wish to pursue operative treatment, despite moderate to severe dysmorphology, and when clinical or radiologic (e.g., “copper-beaten” or “thumbprinting” on 3D CT) indicators of intracranial hypertension are lacking, ICP monitoring could be considered. If ICP is shown to be elevated on lumbar puncture or intracranial monitoring in such children, this provides impetus to proceed with operative correction. It is clear that the presence of papilledema represents only a fraction of the cases of true intracranial hypertension.78,79 However, the invasiveness of ICP testing should be weighed against its utility in operative decision making. Realistic expectations with regard to correction or prevention of developmental delay should be articulated to the parents, as such a result obviously cannot be guaranteed.
The optimal timing of treatment is during infancy, between 6 and 9 months.49,77,80 This is to harness the force of the rapidly growing brain, allowing brain expansion to influence the newly unrestricted calvarial plates. Additionally, the cranial bone in this age group remains malleable and easy to mold and shape, and bony defects, created during calvarial repositioning, are more apt to reossify completely. The infant is also more prepared to endure the anesthetic risk and the hematopoietic nadir has passed by 6 months. Additionally, intervention during this time may result in normalization of the cranial base.52,81 Delay in surgery after 1 year of life is not advocated as it may result in further progression of the dysmporhology, possible secondary compensatory aberrations (including asymmetry of the facial skeleton), and development of neuropsychiatric problems.82 However, some have claimed that surgical delay until after 12 months may carry the benefit of reduced need for secondary revision.83 Proponents of endoscopic techniques prefer intervention even earlier than 6 months, to take further advantage of brain doubling size.
Treatment/surgical technique
Preparation
The anesthesia team should diligently follow hemodynamic parameters and hemoglobin levels throughout the case, with low threshold for transfusion. Systemic administration of antifibrinolytics (e.g., aprotinin or aminocaproic acid) may be beneficial in reducing blood loss and is an ongoing study at our center.84–86 Blood loss is a continual process occurring from the expansive surface area created by the scalp flap, calvarial plates, and dura. In rare instances, acute significant blood loss can occur and, when present, is most frequently related to dural sinus tears in the setting of repeat operation with osseous-dural adhesions. Placement of so-called blocking stitches (running interlocking 2-0 Prolene) on either side of the planned coronal incision and injection of vasoconstrictors (e.g., epinephrine–kenalog mixture) along the incision and planned area of dissection help minimize bleeding from the scalp flap. Blocking sutures are preferred to Rainey clips for the possible reduced incidence of alopecia.87 Similarly, needle point electrocautery is ill advised along the incision because of damage to follicles leaving a wider, hairless scar.88 However, electrocautery is utilized in those cases where epinephrine is contraindicated (e.g., long QT syndrome). Supraperiosteal dissection along the cranium and subperiosteal dissection intraorbitally also diminish blood loss. Vigilant hemostatic control of oozing surfaces by use of bone wax, electrocautery, and bipolar, as indicated, should also be performed.
A coronal incision is required for most cranial vault remodeling procedures. The incision should be placed from ear to ear, overlying the vertex, and attention paid to potential future procedures or presence of existing incisions (e.g., ventricular shunt). The design can be a straight line or zigzag. The scar is most noticeable in the temporal region and a zigzag pattern is preferred at least in this location, and is often continued along the entire length of incision.89 The knife blade should be beveled anteriorly to preserve viable hair follicles, allowing growth through the scar (i.e., trichophytic closure). If an endoscopic approach is pursued, this typically requires two shorter incisions, one posterior and anterior.90 Occasionally an upper-eyelid incision is needed in endoscopic techniques to address the orbital rim position.91
The choice of operation depends on the specific suture fused and the degree of dysmorphology. In general, the technical surgical goals entail: releasing the area of sutural fusion, repositioning the bone in an anatomic but overcorrected location, eliminating secondary compensatory changes, filling in osteotomy gaps with bone dust slurry, and closing the soft tissue relatively tension-free. Open cranial vault remodeling provides necessary access and freedom to accomplish these goals. Comprehensive, open techniques are preferred to treat craniosynostosis with increasing severity of the deformity. The limited approaches (e.g., endoscopic or linear craniectomy) may be applicable for milder skull deformities, certainly not involving more than a single suture, in younger patients (<3–6 months).4 These limited or endoscopic approaches do not reposition hypoplastic bony segments, but rather rely on the expanding brain, alone or coupled with helmet therapy, to correct the skull deformity.90 The frontal region must be addressed in metopic and coronal synostoses and most effectively done in a full-access manner.
Stabilization of repositioned bone segments is best performed with sutures, wires, or resorbable plates and screws. The objective is to create a stable construct but not to restrict brain growth and subsequent skull expansion. Titanium or metallic plates are not utilized in infants undergoing cranial remodeling for fear of transcranial migration with subsequent growth.92–96 Free-floating forehead techniques have been attempted in the past, but, without stability, a permanent deformity may result.97 Obviously, limited or endoscopic techniques do not utilize fixation, as they involve strip suturectomies and/or barrel staves only.
Sagittal synostosis
There are many options for treating sagittal suture fusion. In the neonatal period, when the anteroposterior deformity may be mild, a simple strip craniectomy (sagittal synostectomy), or variation thereof, may be performed. This can be done by either an open or an endoscopic approach. The space created between parietal bones at the vertex may allow the head shape to normalize with brain growth (the fronto-occipital distance shortens and the biparietal distance expands).63 However, a semblance of frontal bossing or occipital prominence can persist over time. Additionally, the medial border of the parietal bones, at the site of the suturectomy, can refuse with recurrence and progression of scaphocephaly noted. This is not an attractive surgical option for infants older than 6 months because: (1) it does not adequately correct the scaphocephaly; and (2) permanent osseous defects can result along the vertex.
The endoscopic approach for treatment of craniosynostosis was introduced by Jimenez and Barone.90 The original cohort included 4 infants all under 3 months of age, with a follow-up period for approximately 1 year. The technique described includes a strip craniectomy with lateral barrel staves. A crucial component of this approach is use of the molding helmet postoperatively. Early results were successful gauging by aesthetic criteria. Additional practitioners have reported on using endoscopic access for treatment of sagittal synostosis, with various modifications.98 It has been demonstrated that, in infants younger than 3–6 months of age, treatment of sagittal synostosis by suturectomy and parasagittal osteotomies can be effective.
If the dysmorphology is more significant, but still minimal frontal prominence, the “pi” procedure can be performed. The pi procedure was first described in 1978 by Jane et al. and produces immediate correction of the fronto-occipital length and biparietal width.99 Technically, it involves two parallel parasagittal ostectomies of the two parietal bones, connected with a transverse ostectomy, located behind the coronal suture and extending to the temporal region (resembling the Greek symbol for pi). The dura is dissected free from the endocranial surface of the frontal bone and the remaining fused sagittal suture, to prevent buckling when reapproximating new bone edges. Displacing the frontal bone posteriorly results in bulging of the brain laterally. These two movements are key to improving the morphology of scaphocephaly. Though probably not more effective than strip craniectomy during the neonatal period, the pi procedure offers the advantage of immediate aesthetic correction of head shape and improved efficacy in older infants (e.g., 8 months).
Moderate to severe scaphocephaly is best corrected by total cranial vault reconstruction, as the more limited techniques described will not adequately correct the dysmorphology100–102 (Fig. 34.3). This is best done between 6 and 9 months and entails excision of the frontal, parietal, and occipital bone plates, which are molded, reshaped, and repositioned (Fig. 34.4). The occipital region is advanced forward and the frontal prominence retropositioned (Fig. 34.5

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


