Six principal problems are encountered using this algorithm. The first is that the separation plane may change as blisters age. Spongiotic microvesicles may move into the stratum corneum as crust. The epidermal blister roofs may become necrotic, not allowing assessment of the original blister plane. However, the basal keratinocytes may retain their columnar appearance in the blister roof (or base), allowing for separation into either suprabasal or sub-basal blister formation. Also, basal keratinocytes are the most melanized cells in the epidermis. Search for the melanized basal keratinocytes (i.e., the basal unit), even if they are necrotic, may allow assessment of the original blister plane. Immunohistochemical stain for type IV collagen is sometimes useful in sublamina densa blistering disorder because type IV collagen may be detectable on the base of subepidermal blister roofs even when the blister roof is necrotic. Some blister roofs may be viable for a week or so; in this case, the viable keratinocytes may become elongated and line the base of the blister roof. Similarly, the reepithelialized subepidermal or suprabasal blister bases are initially lined by flat squamous keratinocytes rather than columnar or cuboidal keratinocytes. When reepithelialization occurs first, there are no normal rete ridges and the flat squamous keratinocytes migrate on a matrix of fibrinogen, not normal papillary dermis. Second, microscopic slit-like spaces occur within the epidermis in the group of clefting diseases—Darier disease, Hailey–Hailey disease, and Grover disease—mimicking true blisters. However, these clefts are small and do not contain fibrin and tissue fluid in the vast majority. Furthermore, this group of disorders only rarely presents clinically with blisters. Third, evaluation of routine histologic preparations may not allow one to accurately assess the specific mechanism of blister formation. This is particularly true in the subepidermal vesiculobullous disorders. Fourth, the cell types infiltrating the lesions in the vesiculobullous diseases change as the lesions age. Fifth, the histologic descriptions of many of the subepidermal blistering disorders are not accompanied by the rigorous immunologic evaluation required today. For example, many cases originally reported as bullous pemphigoid and cicatricial pemphigoid (CP) are now known to be better classified as EB acquisita and linear IgA bullous dermatosis. Many of the subepidermal blistering disorders may strikingly mimic each other clinically, leading to inappropriate diagnosis and making the histologic descriptions in the literature suspect. Finally, in order to rapidly improve the rash and symptomology of the patient, clinicians often embark on therapeutic regimens for the presumed clinical diagnosis. The therapy often consists of topical or systemic steroids and inhibitors of inflammation. Histopathologic examination of a biopsy is only pursued when the therapeutic response is nil or partial, calling into question the clinical diagnosis. Many submit biopsies without notifying the dermatopathologist of the therapy. Such a therapy often suppresses the inflammatory infiltrate, the epidermal response, and other histopathologic changes. The evolution and temporal relationship of histologic changes of dermatitides under various therapies are not sufficiently documented to allow optimal, or perhaps, even accurate diagnostic study. Biopsies should be taken of virgin disease not of therapeutically altered skin if at all possible. If therapy has been instituted, it should be stopped, if possible, at least a week before biopsy.
Principles of Management
Principles of management for bullous disorders are diverse, reflecting their differing etiologies, mechanisms, and sites of predilection. In the sections that follow, the management principles are briefly discussed for each of the major disease entities. In the final section, the management of immunobullous disorders is discussed in a more global manner.
With these points in mind, the specific disorders and groups of disorders will be discussed.
SPONGIOTIC DERMATITIS
Definition and Evaluation
Spongiotic dermatitis may be acute, subacute, or chronic. The process is dynamic, and each specific type of dermatitis may progress from the acute to the chronic phase. Although the term spongiotic dermatitis is occasionally used interchangeably with eczema, the word eczema lacks specific meaning. Spongiosis refers to the accumulation of edema fluid between keratinocytes, in some cases progressing to vesicle or even bulla formation.
In acute spongiotic dermatitis, the stratum corneum is normal and the epidermis is of normal thickness. The degree of spongiosis is variable, extending from slight to marked, with intraepidermal vesiculation in the latter instance. The surrounding keratinocytes may be stellate, surrounded by clear spaces representing the site of fluid collection (Fig. 9-1). Papillary dermal edema is present, correlating with the degree of spongiosis. A lymphohistiocytic infiltrate is present around the superficial plexus, with exocytosis of lymphocytes into spongiotic foci.
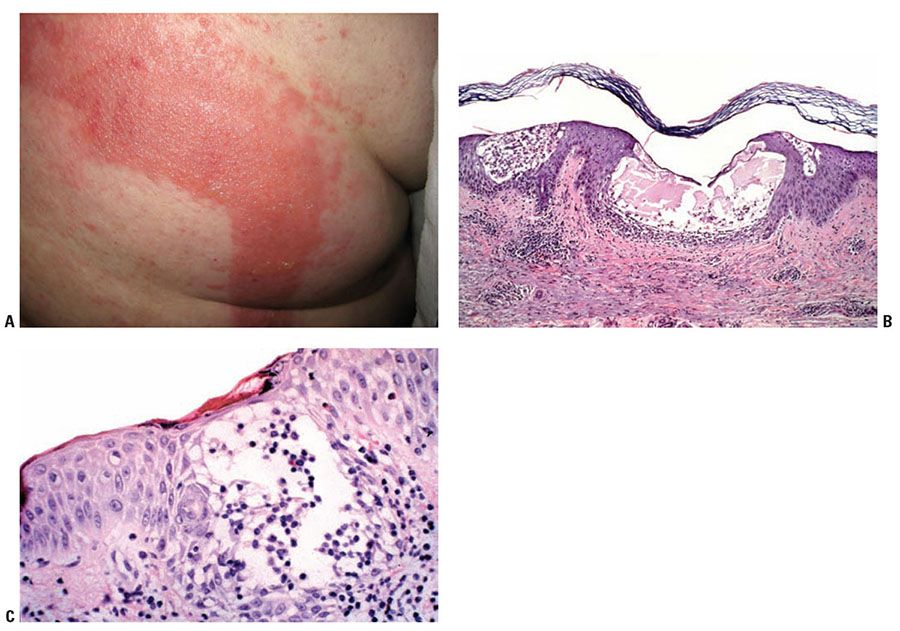
Figure 9-1 Acute dermatitis: allergic contact dermatitis. A: Geometric erythematous plaque with overlying vesicles on the back of a patient with allergic contact dermatitis to adhesive tape. B: Spongiosis with vesicles containing fluid and inflammatory cells. C: Exocytosis of lymphocytes and eosinophils into the spongiotic epidermis.
In subacute spongiotic dermatitis, there is usually mild-to-moderate spongiosis, occasionally with microvesiculation. The epidermis is moderately acanthotic. The parakeratotic stratum corneum may contain aggregates of coagulated plasma and scattered lymphocytes and neutrophils, forming a crust (Fig. 9-2). There is a superficial perivascular lymphohistiocytic infiltrate, which is less prominent than in the acute phase. Impetiginization by gram-positive cocci (Staphylococci and Streptococci) may lead to neutrophilic crust.
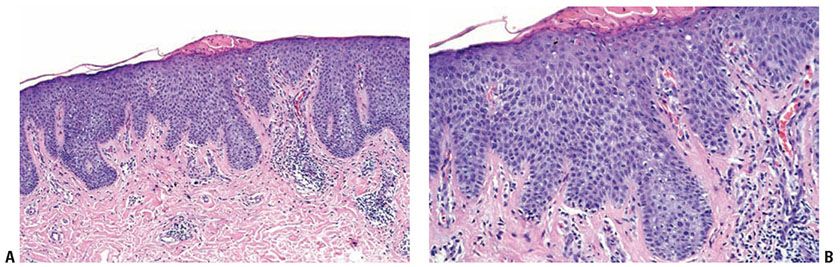
Figure 9-2 Subacute spongiotic dermatitis: nummular dermatitis. A: There is irregular acanthosis, spongiosis, and superficial perivascular inflammatory infiltrate. Parakeratosis containing plasma is also present. B: Spongiosis is accompanied by exocytosis of inflammatory cells.
In chronic dermatitis, there is hyperkeratosis with areas of parakeratosis, often wedge-shaped hypergranulosis, and moderate to marked acanthosis. Spongiosis may be present focally, often minimal in degree. The inflammatory infiltrate is often sparse, and papillary dermal fibrosis may be a prominent feature. The degree of spongiosis and quantity of the infiltrate reflect the present activity of the underlying dermatitis. The other changes reflect lichen simplex chronicus (Fig. 9-3) with its vertical streaking of thick papillary dermal collagen.
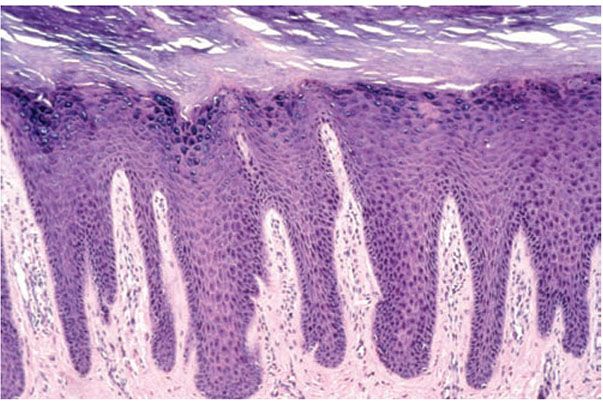
Figure 9-3 Lichen simplex chronicus. There is hyperkeratosis, hypergranulosis, and irregular psoriasiform acanthosis with minimal spongiosis. Vertically oriented collagen in the papillary dermis is characteristic.
Specific Types of Spongiotic Dermatitis
Allergic Contact Dermatitis
Clinical Summary. The prototype of acute spongiotic dermatitis is allergic contact dermatitis, secondary to exposure to poison ivy. Usually between 24 and 72 hours after exposure to the antigen, the patient develops pruritic, edematous, erythematous papules and plaques and, in some cases, vesicles. Linear papules and vesicles are common in allergic contact dermatitis to poison ivy, reflecting the points of contact between the plants or the antigen-contaminated hands and the skin. Other common causes of allergic contact dermatitis (Fig. 9-1A) include nickel, paraphenylenediamine, rubber compounds, fragrances, and preservatives in cosmetics. The degree of histologic alterations to these antigens may be less striking than that secondary to poison ivy.
Histopathology. Early lesions are acute spongiotic dermatitis. If vesicles develop, they may contain clusters of Langerhans cells, some flask shaped. There is a superficial dermal infiltrate of lymphocytes, macrophages, and Langerhans cells with accentuation around the small vessels. Eosinophils may be present in the dermal infiltrate as well as within areas of spongiosis (Fig. 9-1). In patients with continued exposure to the antigen, the biopsy may show a subacute dermatitis or, later, a chronic spongiotic dermatitis, often with lichen simplex chronicus due to rubbing.
The histopathologic features, as outlined, are idealized, and in any instance, reflect the character of the hapten, its resultant hapten-protein complex, and the particular immune response to that antigen. Some antigens may not produce but minimal changes.
Pathogenesis. Allergic contact dermatitis represents a type IV cell-mediated delayed hypersensitivity reaction. The immunologic reaction consists of an afferent limb, the sensitization or induction phase, and an efferent limb, the elicitation phase. The allergen is usually of low molecular weight (a hapten) and lipid soluble. After penetrating the skin, the hapten binds to a carrier protein to form a complete antigen, which is processed by antigen-presenting cells, principally the epidermal Langerhans cells, and possibly other cutaneous dendritic cells. These cells then migrate to the draining lymph nodes, where they present the antigen to T lymphocytes. The process triggers antigen-specific commitment and production of memory and effector T cells. Following subsequent exposure of the sensitized subject to the same allergen, an accelerated and more aggressive secondary immune response will be elicited. Both CD4+ and CD8+ subtypes of T lymphocytes participate in contact hypersensitivity reactions (1). The homing of lymphocytes to the antigen in exposed skin requires various cell adhesion molecules, such as lymphocyte function-associated antigen-1 (LFA-1) (2). Lymphocytes liberate various cytokines in the affected area of skin, including interleukins, IFN-γ, TNF-α, leading to a further influx of inflammatory cells, particularly nonsensitized lymphocytes and eosinophils. Chemokines appear to be crucial regulators of both induction and expression of allergic contact dermatitis. In contrast to lipid-soluble hapten, metal ions activate inflammatory response by different mechanisms. Nickel, for example, directly activates Toll-like receptor 4 (TLR4) and generates a signal that promotes inflammation (3).
Principles of Management. Contact dermatitis is best managed through avoidance; patch testing can offer patients a list of causative allergens and databases can provide registries of safe-to-use products. Active lesions often require treatment with topical corticosteroids; widespread or severe eruptions may warrant systemic corticosteroid therapy.
Irritant Contact Dermatitis
Clinical Summary. This inflammatory reaction occurs after exposure to an irritant, a toxic compound that causes a reaction in most individuals who come into contact with it. Common irritants include alkalis, such as soaps, detergents, lye, and ammonia-containing compounds. The irritant response is determined by the type of chemical, its concentration, the mode of exposure, the body site, the barrier function locally, and the age of the patient. Atopy is a predisposing factor. The clinical morphology varies. Sometimes it is indistinguishable from allergic contact dermatitis.
Chemical burns, usually caused by strong alkalis and acids, lead to immediate painful erythema progressing to vesiculation, necrosis, and, if severe, ulceration. Acute irritant reactions produce a monomorphic picture with scaling, redness, vesicles, pustules, or erosions, and are caused by such mild irritants as detergents and water with additives. Agents producing this pattern include tretinoin, benzalkonium chloride, dithranol, adhesive tapes, and cosmetics. Dryness, chapping, and absence of vesicles characterize chronic irritant contact dermatitis. This reaction pattern is produced by repetitive contact to water, detergent, and solvents.
Histopathology. The histologic picture varies from extensive ulceration, to simply diffuse hyperkeratosis or parakeratosis with congestion and ectasia, to a spongiotic pattern essentially identical to allergic contact dermatitis. The variable features reflect the protean factors discussed above. Some correlations are worthy of note. In some instances, there is significant necrosis with nuclear karyorrhexis and cytoplasmic pallor (Bandmann achromia). In severe reactions; the necrosis may extend into the dermis. Some irritants such as cantharidin and trichloroethylene produce acantholysis and neutrophilic infiltration in the epidermis (Fig. 9-4) (4). Other contactants may specifically target vascular endothelium. If ulcers are present, the exudate may produce an irritant effect. Some reactions, however, may be entirely spongiotic, such as those due to weak irritants, strong irritants in low concentration, and those in the “irritable skin syndrome.” Because of these observations and those made in positive patch test sites, routine histopathologic changes do not reliably separate irritant from allergic contact reactions, although necrosis, neutrophilic infiltration, and acantholysis are more frequent in the former. In the recovery phase of irritant dermatitis, mild epidermal hyperplasia, sometimes psoriasiform, is often present. Psoriasiform hyperplasia may develop in chronic irritant reactions. In other words, most common irritants lead to subacute and chronic spongiotic dermatitis histologic features identical to those of allergic contact reactions.
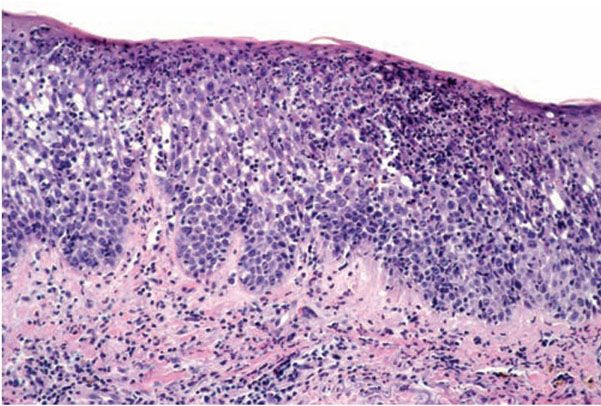
Figure 9-4 Irritant dermatitis. There is superficial epidermal necrosis together with spongiosis and infiltrate of neutrophils.
Pathogenesis. The pathogenesis at the cellular and molecular levels of irritant contact dermatitis is not completely understood. Mechanisms of damage are variable with different irritants; these include keratin denaturation, damage of the permeability barrier through removal of surface lipid and water-holding substances, disruption of cell membranes and direct cytotoxic effects. Recent studies indicate that very complex reaction patterns involving immunologic components occur in the irritant response. There are surprisingly more similarities than differences between irritant contact dermatitis and allergic contact dermatitis in morphology, clinical manifestation, chemokine expression and involvement of T cells (5–7).
Principles of Management. Avoidance is essential. Often irritant dermatitis can be managed through use of appropriate protective equipment (such as cotton-lined rubber gloves when doing dishes) and aggressive emollient use.
Dyshidrotic Dermatitis
Clinical Summary. This entity is characterized by recurrent, severely pruritic, tense vesicles that classically involve the lateral aspects of the fingers and, in some cases, the toes. Episodes may be precipitated by infections, “id” reactions, contact reactions, and emotional stress, oftentimes associated with atopic dermatitis. In chronic cases, there may be more extensive involvement of the palms and soles. Although the eruption develops acutely, it may become chronic with erythema, lichenification, and fissuring. Secondary impetiginization is common.
Histopathology. Spongiosis and intraepidermal vesiculation occur in acute lesions (Fig. 9-5). There is a superficial perivascular lymphohistiocytic infiltrate with exocytosis of lymphocytes into spongiotic zones. The infiltration is usually mild. In acute lesions, the compact, thickened stratum corneum of acral skin remains intact, and the epidermal thickness is normal. With chronicity, spongiosis diminishes; acanthosis and parakeratosis predominate with variably crusting. Difficulty in differential diagnosis with pustular psoriasis may occur because of the formation of vesiculopustules in older lesions. A periodic acid–Schiff (PAS) stain should always be performed on vesicular lesions of the palms and soles, because tinea manus/pedis may mimic dyshidrotic dermatitis histologically.
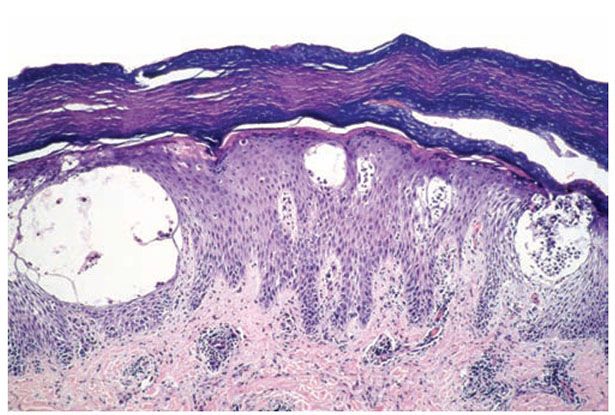
Figure 9-5 Dyshidrotic dermatitis. In acral skin, large intraepidermal vesicles are held intact by the thick stratum corneum.
Principles of Management. Maintaining a healthy skin barrier with avoidance of irritants and aggressive moisturization is essential; flares of disease may be managed with topical corticosteroids.
Autoeczematization or “Id” Reaction
Clinical Summary. A sudden generalized or localized vesicular dermatitis developing in association with a defined local dermatitis or infection is known as an “id” reaction. The lesions are pinhead-sized, acuminate or flat-topped, papules. The patient most often has bullous tinea pedis or kerion, thus the name, “dermatophytid.” In some cases, patients have a severe localized dermatitis, such as stasis dermatitis; with subsequent development of widespread papulovesicular lesions (8). Other forms of id reaction exist.
Histopathology. The features are those of acute spongiotic dermatitis, often with micro- or macrovesiculation. Eosinophils may be present. There is often some edema of the superficial papillary dermis with some large lymphocytes (presumably activated) in the upper dermis (9). The infiltrating cells are T cells—those within the epidermis are principally CD8+ and those within the dermis are principally CD4+.
Pathogenesis. Id reactions have been postulated to represent hypersensitivity dermatitis to an autoantigen; however, this has never been proven. Several other possibilities exist. First, the reaction may represent a conditioned hyperirritability or responsiveness precipitated by the local infection or dermatitis. Acute local chemical irritation may lower the threshold for an irritant reaction to the same chemical at remote sites. Second, localized dermatitis leads to local production of cytokines by keratinocytes. If the cytokines are hematogenously disseminated, they may lead to distant skin hyperirritability. Circulating activated T lymphocytes may play a role in “id” reaction associated with allergic contact dermatitis. Lastly, dissemination of antigen (but not of whole infectious agents) from the local site may occur with then a secondary response developing at a site of distant deposition. Microbial antigens have been identified in an id reaction in a patient with tuberculoid leprosy (10).
Principles of Management. “Id” reactions are best managed by calming the inciting localized eruption, and treating the inflamed skin with topical steroids and emollients.
Photoallergic Dermatitis
Clinical Summary. Photoallergy is increased reactivity of skin to ultraviolet and visible light and is brought about by a chemical agent on an immunologic basis (11). The eruption of photoallergic dermatitis may be due to topical application of, or oral ingestion of, a photosensitizing agent, resulting in a photocontact dermatitis or photodrug eruption, respectively. Agents that may elicit photocontact dermatitis include soaps and cleansers (containing halogenated salicylanilide), perfumes (such as Musk ambrette), topical sulfonamides, sunscreens, benzocaine, and diphenhydramine. Common causes of photodrug eruption include thiazide diuretics, oral hypoglycemics, and phenothiazines. The eruption is pruritic and composed of erythematous papules and confluent plaques on sun-exposed skin, usually the face, dorsal aspect of the arms, and the “V” of the neck.
Histopathology. The features are similar to that of an acute allergic contact dermatitis, showing variable spongiosis, in some cases with vesiculation, and a superficial perivascular lymphohistiocytic infiltrate with exocytosis. A deeper perivascular infiltrate and eosinophils are more common in photoallergic dermatitis induced by systemic ingestion of medications. With chronic exposure to the antigen, there is progression to chronic dermatitis, with diminished spongiosis, less intense inflammation, and more acanthosis.
Pathogenesis. Phototoxic reactions are “sunburn” reactions with epidermal apoptosis and necrosis with intraepidermal to subepidermal blisters—the changes paralleling the degree of damage. Neutrophils may be prominent.
Principles of Management. A thorough history for exposures, potentially including patch testing or photo-patch testing may reveal the causative agents and help with avoidance. Sun protection may be beneficial (though sunscreens may be the causative culprit in some cases). Topical steroids may offer some benefit in select cases.
Nummular Dermatitis
Clinical Summary. The eruption is characterized by intensely pruritic, coin-shaped (nummular), erythematous, scaly, crusted plaques. The lesions tend to develop on the extensor surfaces of the extremities, and are often mistaken for “ringworm,” or tinea corporis.
Histopathology. Nummular dermatitis is the prototype of subacute spongiotic dermatitis (Fig. 9-2). There is mild-to-moderate spongiosis, usually without vesiculation. Irregular acanthosis with some exocytosis of inflammatory cells is usually present. The parakeratotic stratum corneum contains aggregates of coagulated plasma, forming a crust. Mild papillary dermal edema and vascular dilatation may be present. There is a superficial perivascular infiltrate of lymphocytes, some eosinophils, and occasional neutrophils.
Pathogenesis. The etiology of this entity is unknown. The routine and ultrastructural changes in nummular dermatitis resemble those of contact dermatitis. Intercellular edema is the most conspicuous finding. There is shearing and loss of desmosomes as spongiosis becomes marked.
Principles of Management. Nummular dermatitis often requires high-potency topical steroids to achieve relief.
Atopic Dermatitis
Clinical Summary. The eruption is characterized by areas of severe pruritus, erythema, scaling, and excoriation, and with chronicity, lichenification. Acute lesions of atopic dermatitis begin as erythematous papules and serous exudates. Secondary lesions include excoriations and crusted erosions due to scratching. Subacute lesions appear as erythematous scaling papules and plaques. If the itch and rash progress uncontrolled, chronic lichenified atopic dermatitis develops featuring accentuated skin markings and hyperpigmentation. Most patients are diagnosed in childhood; approximately one-third of cases are diagnosed before 1 year of age (12). There is a female predominance, and many patients have other atopic disorders such as allergic rhinitis or asthma. Increased incidence of contact dermatitis and susceptibility to cutaneous infection is also noted among patients with atopic dermatitis. In infants, the lesions predominate on the face and extensor surfaces of the extremities, but they later affect the flexural surfaces. The classically involved sites in older children and adults are the popliteal and antecubital fossae and the sides of the neck. Secondary bacterial infection is common.
Histopathology. In early phases, there is mild spongiosis, exocytosis of lymphocytes, and parakeratosis. Lymphocytes and scattered histiocytes are present around the superficial vascular plexus. In long-standing lesions, the rete ridges are regularly elongated, with less prominent spongiosis and cellular infiltrate. Hyperkeratosis and wedge-shaped hypergranulosis with areas of parakeratosis develop. There appears to be an increased number of small vessels with thickened walls. Eosinophils are less conspicuous than in allergic contact or nummular dermatitis. With time, the changes may be those of lichen simplex chronicus.
Pathogenesis. The current understanding of the disease remains incomplete. Atopic dermatitis develops as a result of complex interactions of genetic, environmental, and immunologic factors. Both abnormalities of immune regulation and barrier functions of the epidermis are involved.
The immune factors include the differentiation of helper T cells, increased life span of eosinophils, multiple roles of IgE, the pattern of local cytokine expression, infectious agents, and superantigens (13). The initiation of atopic dermatitis appears to be driven by allergen-induced activation of type 2 T helper cells (TH2 cells), leading to increased levels of interleukin-4, -10, and -13. Elevated IgE ensues. IgE contributes to the inflammatory cell infiltrate by several mechanisms, including immediate/late phase reaction, allergen presentation by IgE-bearing Langerhans cells and allergen-induced activation of IgE-bearing macrophages.
The mononuclear cell infiltrate in the lesions probably reflects a combination of both IgE-dependent mast cell/basophil degranulation and TH2 cell-mediated responses elicited during acute exposure to allergens, including ingestants, inhalants, or contact aeroallergens such as human dander, grass pollens, and house dust mites (14).
The pattern of local cytokine expression plays an important role in modulating tissue inflammation and depends on the activity and/or duration of the lesion. Acute skin inflammation is associated with a predominance of interleukin (IL)-4 and IL-13 expression, and little INF-γ. But in chronic lesions, there are increased INF-γ-producing cells (15,16).
Abnormal epidermal barrier function is a well-recognized clinical association of atopic dermatitis. Filaggrin gene mutations, which result in impairment in barrier function and increased ease of allergen presentation to epidermal dendritic cells, may potentiate contact reactions (17).
No specific single gene is a unique marker for atopic dermatitis. Genome screens of families with atopic dermatitis have implicated chromosomal regions that overlap with other skin diseases, including inflammatory and autoimmune skin diseases (18). The dysfunctional skin barrier genes, which include loss of functional filaggrin on chromosome 1q21 (19), increase of protease activity and lack of protease inhibitor, predispose the patients to the harmful effects of environmental agents (20). Chromosome 5q31 contains the IL-4 gene cluster family. Because IL-4 is central to the induction of IgE synthesis by B cells, it has been suggested that polymorphisms in the chromosome 5q31 region may be linked to the gene controlling total serum IgE in atopic individuals. Another possible candidate gene found on chromosome 11q13 is the high-affinity IgE receptor (21,22).
Principles of Management. Patients should practice good bathing practices to improve epidermal barrier function. Lukewarm baths, moisturizing soaps, avoiding harsh abrasive scrubs, and extensive moisturization with thick, bland emollients is essential. Inflammation often requires topical corticosteroids. Patients should be evaluated for superinfection including both bacterial and sometimes viral infections.
Lichen Simplex Chronicus
Clinical Summary. The vast majority of patients with pruritus who chronically rub the skin may develop lichen simplex chronicus. It often develops in the setting of atopic dermatitis or allergic contact dermatitis. The lesions are pruritic, thick plaques often with excoriation, in which the normal skin markings are accentuated, the latter finding known as lichenification. Uncommonly, some patients develop macular or lichen amyloidosis.
Histopathology. Lichen simplex chronicus is the prototype for chronic dermatitis (Fig. 9-3). There is hyperkeratosis interspersed with areas of parakeratosis, acanthosis with irregular elongation of the rete ridges, wedge-shaped hypergranulosis, and broadening of the dermal papillae. Slight spongiosis may be observed, but vesiculation is absent. Minimal papillomatosis is sometimes present. Excoriations, which are punctate ulcerations lined by necrotic superficial papillary dermis, fibrin, and neutrophils, are often present; however, they are often present in many pruritic dermatoses. There may be a sparse superficial perivascular infiltrate without exocytosis. In the papillary dermis, there are an increased number of fibroblasts and vertically-oriented collagen bundles. As rubbing increases in intensity and chronicity, epidermal hyperplasia becomes more florid, and the fibrosis more marked.
Pathogenesis. Features associated with a specific dermatitis may allow identification of the underlying cause.
Principles of Management. Management of lichen simplex chronicus requires mid- to high-potency topical steroids, often in an ointment form, and aggressive skin moisturization.
Seborrheic Dermatitis
Clinical Summary. Clinically, patients develop erythema and greasy scale on the scalp, ears, eyebrows, nasolabial areas, and central chest. Rarely, patients with seborrheic dermatitis develop generalized lesions. In infants, the scalp (“cradle cap”), face, and diaper areas are often involved. It is said to be a rare cause of erythroderma. Seborrheic dermatitis is seen with increased frequency in patients with Parkinson disease, epilepsy, congestive heart failure, chronic alcoholism, zinc deficiency, and HIV infection. Patients with HIV infection often have severe refractory disease and atypical distribution (23).
Histopathology. The histopathologic features are a combination of those observed in psoriasis and spongiotic dermatitis. Mild cases may exhibit only slight subacute spongiotic dermatitis. The stratum corneum contains focal parakeratosis, with a predilection for the follicular ostia, a finding known as “shoulder parakeratosis” (Fig. 9-6). Occasional pyknotic neutrophils are present within parakeratotic foci (neutrophilic parakeratosis) sometimes with fluid (neutrophilic crust). There is moderate acanthosis with regular elongation of the rete ridges, mild spongiosis, and focal exocytosis of lymphocytes. The dermis contains a sparse mononuclear cell infiltrate. In HIV-infected patients, the epidermis contains apoptotic keratinocytes, and the dermal infiltrate usually contains plasma cells.
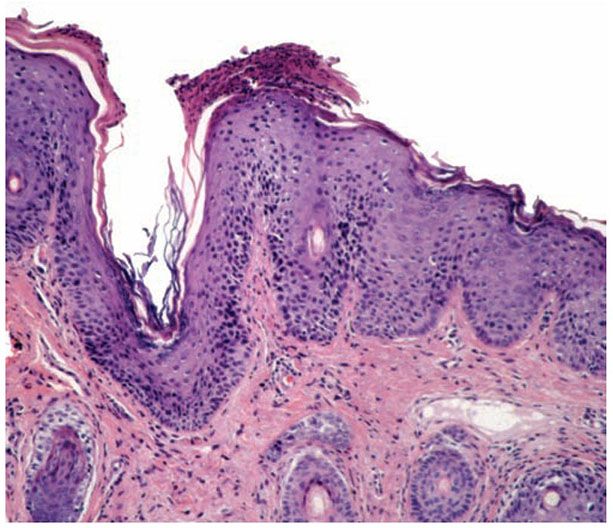
Figure 9-6 Seborrheic dermatitis. There is neutrophilic parakeratosis on the follicular ostial shoulder. The epidermis is hyperplastic. Spongiosis is minimal in this biopsy.
Pathogenesis. The pathogenesis is unknown; the role of Malassezia sp. (Pityrosporum) in the etiology is controversial even though many patients have a good response to oral or topical ketoconazole (24,25).
Principles of Management. Seborrheic dermatitis may improve with antidandruff shampoos available over the counter. Some patients benefit from ketoconazole 2% shampoo and/or cream. Actively inflamed, very red skin may improve faster with low potency topical corticosteroids.
Stasis Dermatitis
Clinical Summary. Patients with long-standing venous insufficiency and lower extremity edema may develop pruritic, erythematous, scaly papules and plaques on the lower legs, often in association with brown pigmentation and hair loss. Ulceration is a frequent complication of long-standing stasis dermatitis.
Histopathology. The variable epidermis may be hyperkeratotic with focal parakeratosis, acanthotic to atrophic and with focal spongiosis. There is proliferation of small blood vessels in the papillary dermis, forming lobular aggregates (glomeruloid proliferation). The proliferation may be florid, mimicking Kaposi sarcoma (acroangiodermatitis) (26). There is a superficial perivascular lymphocytic infiltrate that surrounds thickened capillaries and venules. The reticular dermis is often fibrotic. Extravasated erythrocytes and hemosiderin are usually present superficially, but they may be identified about the deep vascular plexus as well. Fibrin thrombi may be observed in the small vessels, likely reflecting flow disruption and anoxia. They do not indicate a concurrent coagulopathy. Endothelial necrosis and neutrophils may be present as well reflecting similar changes and do not indicate a leukocytoclastic vasculitis.
Principles of Management. Stasis dermatitis requires optimization of the total body fluid status, elevation of the legs as much as possible, and gentle compression therapy. Actively inflamed skin may benefit from a short course of topical steroids followed by aggressive emollient use.
Differential Diagnosis of Spongiotic Dermatitis
Acute spongiotic dermatitis may be seen histologically in pityriasis rosea, guttate parapsoriasis, digitate dermatitis, spongiotic drug eruptions, arthropod bite reactions, and dermatophyte infections. Lesions of pityriasis rosea contain periodic discontiguous suprapapillary spongiosis, exocytosis of lymphocytes, and overlying mounds of (lenticular) parakeratosis. The spongiosis varies from micro- to macrovesicular. In addition, extravasated erythrocytes may be present in the involved papillae and overlying epidermis. Guttate parapsoriasis is similar to pityriasis rosea, but it lacks the intraepidermal erythrocytes and has less lymphohistiocytic infiltrate. Spongiotic drug eruptions tend to have a deeper infiltrate with eosinophils than other forms of acute spongiotic dermatitis. They usually do not vesiculate. In arthropod bite reactions, the epidermal changes are focal, corresponding to the site of attack, and the infiltrate is wedge-shaped. Bite or sting parts are often not found.
Pityriasis lichenoides, dermatophyte infection, or an arthropod bite reaction may show histopathologic features similar to subacute spongiotic dermatitis. In pityriasis lichenoides et varioliformis acuta, the lesions are sharply circumscribed. Apoptotic keratinocytes, dry neutrophil-rich scale, bandlike interface dermatitis, areas of epidermal necrosis, and a deeper perivascular infiltrate are present. Dermatophyte infections characteristically have neutrophils within the stratum corneum, along with fungal hyphae (Gottlieb sign); however, PAS stain is often necessary to identify the fungal hyphae.
Conditions that have features of a chronic dermatitis include pellagra and other nutritional deficiencies, mycosis fungoides, and psoriasis, particularly if altered by therapy. In the nutritional deficiencies, upper epidermal pallor, necrosis, and neutrophilic infiltrate are characteristic. In mycosis fungoides, psoriasiform epidermal hyperplasia may be present, but the lymphocytes may have nuclear atypia and cerebriform morphology with epidermotropism, often tagging in the basal unit most frequently in the absence of spongiosis. Lesions of psoriasis may resemble lichen simplex chronicus, but thinning of the suprapapillary plates, confluent parakeratosis, neutrophils within the stratum corneum and upper epidermis, and dilated tortuous capillaries juxtaposed to the suprapapillary epidermis are distinctive.
Other Disorders with Spongiotic Dermatitis
Erythroderma and Generalized Exfoliative Dermatitis
Clinical Summary. Erythroderma is characterized by generalized erythema over more than 70% to 80% of the body surface area, accompanied by scale, often in association with fever. The eruption is nonspecific and may be caused by a various underlying conditions. In one study, 74.4% were associated with a preexisting dermatosis, 14.6% were idiopathic, 5.5% were related to drugs and malignancy (27). Psoriasis is the most common preexisting dermatosis; others include spongiotic “eczematous” dermatitis, pityriasis rubra pilaris, photosensitivity syndromes, and, rarely, scabies infestation, dermatophytosis, dermatomyositis, acute graft-versus-host, pemphigus foliaceus, and even bullous pemphigoid. Many drugs have been incriminated, such as phenytoin, thiazides, nonsteroidal anti-inflammatory drugs (NSAIDS), and recombinant cytokines; erythrodermatous drug eruptions should be considered severe and patients should be evaluated for signs of systemic inflammation. A small percentage of erythroderma is associated with lymphoma, either paraneoplastic or more frequently cutaneous T-cell lymphoma, in the form of Sézary syndrome or erythrodermic mycosis fungoides.
Histopathology. A careful search for histologic features of any of the above listed etiologies must be undertaken; however, the nature of the underlying dermatosis is not always discernible in the erythrodermic phase, and the changes are often those of nonspecific subacute spongiotic dermatitis. Erythrodermic lesions associated with underlying psoriasis (28) resemble early lesions of psoriasis with only mild epidermal hyperplasia, mild spongiosis, mounds of parakeratosis with a few neutrophils, and red cell extravasation in the papillary dermis. Blood vessels in the upper dermis are usually dilated. In cases of erythroderma related to mycosis fungoides, atypical cells with cerebriform nuclei are present in the infiltrate. Eosinophils may be present. Drug-related cases might simulate mycosis fungoides with exocytosis, lymphocytic atypia (lymphomatoid), and presence of eosinophils. The presence of rare apoptotic keratinocytes may be a clue to the drug etiology.
The histopathologic findings are helpful in establishing the etiologic diagnosis in only about 40% of cases (29). Repeat biopsies and hematologic studies at regular intervals are recommended in patients without definitive diagnosis.
Principles of Management. The underlying cause of the erythroderma must be identified and treatment tailored toward that condition. Generally patients require supportive care with fluid and electrolyte support. Erythrodermic patients are at risk for secondary infection and bacteremia, high output heart failure, and significant metabolic disturbances. Aggressive topical care, often with wet wraps or baths, thick layers of mid-potency topical steroids, and a protective wrap such as a “sauna suit” may lead to rapid clinical improvement while the underlying cause is being evaluated.
Miliaria
Miliaria develops when sweating is associated with obstruction of the intraepidermal sweat duct. There are three types: miliaria crystallina, miliaria rubra, and miliaria profunda.
Clinical Summary. Miliaria crystallina develops when the sweat duct is obstructed within the stratum corneum. Asymptomatic, small, superficial, noninflammatory vesicles resembling dewdrops develop, mainly on the trunk, after severe sunburn or with profuse sweating during a febrile illness. The vesicles rapidly subside when sweating ceases or the horny layer overlying the vesicles exfoliates. In some cases, the eruption may be present at birth (30).
Miliaria rubra (prickly heat) ensues when the sweat duct is obstructed within the deeper layers of the epidermis. It generally develops during and after excessive sweating in skin covered by clothing. It may also occur after prolonged covering of the skin by occlusive polyethylene wraps. Anhidrosis and heat intolerance result, particularly when the trunk is occluded. The lesions consist of pruritic small papulovesicles surrounded by erythema. Pustules may develop.
Miliaria profunda usually develops after recurrent episodes of miliaria rubra, particularly in tropical climates. The sweat duct is occluded at the dermal–epidermal junction. Lesions are nonpruritic, flesh-colored papules and can result in widespread anhidrosis.
Histopathology. In miliaria crystallina, one observes intracorneal or subcorneal vesicles that are in continuity with the underlying sweat duct. A sparse to moderate infiltrate of neutrophils is seen at the periphery of the vesicle. The surrounding epidermis is spongiotic, and there is papillary dermal edema and sparse superficial perivascular inflammation.
In miliaria rubra, spongiotic vesicles are found in the stratum malpighii. Serial sectioning shows these vesicles to be in continuity with a sweat duct. Periductal lymphocyte infiltration and spongiosis are seen, as is an infiltrate in the underlying dermis (Fig. 9-7). In many instances, the distal, intraepidermal sweat duct is filled with amorphous “casts” that are PAS-positive and diastase-resistant.
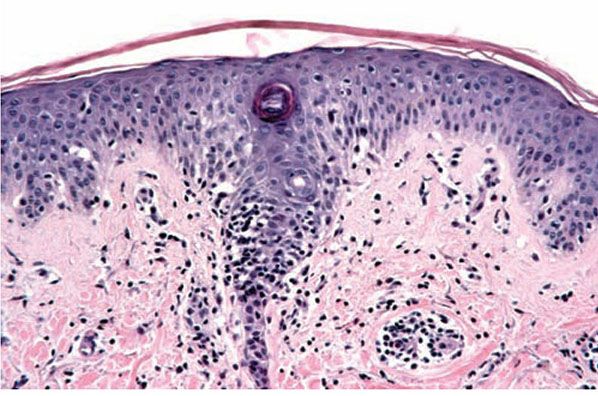
Figure 9-7 Miliaria rubra. There is spongiosis about and within the epidermal eccrine duct. Lymphocytes are present in the areas of spongiosis.
In miliaria profunda, the features are similar to those of miliaria rubra but the inflammatory changes involve the lower epidermis and superficial dermis.
Pathogenesis. In miliaria crystallina, the obstruction of the sweat duct within the stratum corneum is caused either by mild damage to the epidermis from a preceding sunburn or by excessive hydration of the stratum corneum. In neonates, excess hydration of the stratum corneum in utero in combination with immature eccrine ducts may cause swelling of the ductal epithelial cells and occlusion of the duct (31).
In miliaria rubra, increased environmental temperature plays an important role (31). Aerobic bacteria are thought to contribute to the obstruction of the acrosyringium. In favor of this view are the frequent presence of Staphylococcus aureus within the sweat ducts, and the fact that the development of miliaria rubra under an occlusive polyethylene film can be prevented by application of antibacterial solutions.
The PAS-positive, diastase-resistant, amorphous plug within the acrosyringium may occur as a result of injury to the luminal cells, inflammation, and ductal and periductal spongiosis. These changes occur after 48 hours of occlusion and resolve after 14 days, when old, damaged stratum corneum is desquamated. Tape stripping can restore sweating, which supports a reversible, high-level blockage.
Principles of Management. Most miliaria will improve when patients are more mobile and their rooms and skin are cooled off. Some patients benefit from either low- or mid-potency topical steroids or in some cases topical antimicrobial agents.
Immunologic Deficiency Diseases Associated with Spongiotic Dermatitis
Familial Leiner Disease
In this syndrome, infants develop generalized seborrheic dermatitis, severe diarrhea, recurrent local and systemic infections, and marked wasting. Death is usually due to septicemia. A dysfunction of the fifth component of complement (C5) exists, in addition to other cellular and humoral immune defects.
Hyperimmunoglobulin E Syndrome (Job Syndrome)
In this disease, patients have recurrent pyogenic infections, atopic dermatitis, extreme elevation of serum IgE, and defective neutrophil chemotaxis due to decreased production of interferon-gamma (IFN-γ) (32). The infectious complications include recurrent cold staphylococcal abscesses, furunculosis, otitis, sinusitis, and staphylococcal pneumonia.
Wiscott–Aldrich Syndrome
This X-linked recessive disorder affects males and is characterized by recurrent, systemic bacterial and viral infections, purpura due to thrombocytopenia, and an atopic dermatitis-like eruption because of progressive depletion of nodal and circulating T cells; death due to infection or lymphoma ensues in the first decade of life.
Chronic Granulomatous Disease
This X-linked recessive disorder affects males, beginning in infancy with perioral dermatitis. The dermatitis often progresses to granulomatous lesions accompanied by cervical adenitis. Suppurative and granulomatous infections develop in the skin, lungs, bone, and liver, most commonly due to S. aureus. Death occurs in most cases in childhood or adolescence. The defect lies in decreased capacity of neutrophils to generate hydrogen peroxide and kill catalase-positive bacteria and fungi. In vitro, neutrophils are unable to reduce nitroblue tetrazolium dye after phagocytosis, a useful screening test for the disease.
PEMPHIGUS GROUP
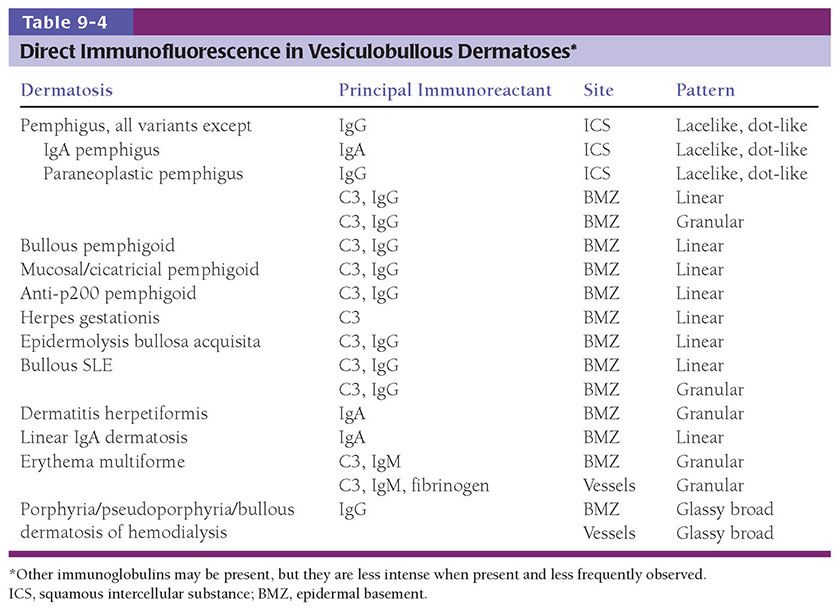
As first demonstrated in 1943 acantholysis is the characteristic feature of the bullae of pemphigus. The acantholysis results from in vivo bound antibodies first discovered in 1964 (33). Pemphigus is now characterized as autoimmune blistering disease presenting clinically with flaccid intraepithelial blisters, erosions and ulcerations of the skin and mucous membranes, histologically with acantholysis and immunologically with in vivo bound and circulating autoantibodies against the cell membrane components of keratinocytes important in cell–cell adhesion. Circulating autoantibodies are present in almost 100% of patients with active disease and their titre usually correlates with disease activity. These antibodies are demonstrable by direct immunofluorescence (DIF) testing of skin and indirect immunofluorescence (IIF) testing of serum. The use of these techniques is now the accepted practice in the diagnosis of all antibody-mediated primary vesiculobullous disorders (see Table 9-4). Pemphigus can be divided into five types: (a) pemphigus vulgaris, with its reactive state, pemphigus vegetans; (b) pemphigus foliaceus, with its lupus-like variant, pemphigus erythematosus, and its endemic variant, Fogo selvagem; (c) drug-induced pemphigus; (d) IgA pemphigus; and (e) paraneoplastic pemphigus (PNP).
Pathophysiology of Pemphigus
The target antigens of pemphigus (Table 9-5) are located in the desmosomes, the most prominent adhesion junctions in stratified squamous epithelium. The desmosome complex contains desmogleins and desmocollins as transmembrane constituents, and plakoglobin, plakophilin, and desmoplakin as cytoplasmic constituents (Fig. 9-8). Desmogleins and desmocollins are members of the cadherin supergene family (34,35), a group of calcium-dependent proteins that play an important role in the formation and maintenance of tissue integrity. The cadherin molecules form dimers as their functional unit, with the extracellular domain from one cell binding to an opposing cell. The cytoplasmic domain of the cadherin associates with plakoglobin, which links intermediate filaments (i.e., keratins) to the desmosome through desmoplakin. The extracellular epitopes of the desmogleins and desmocollins are targets of the pathogenic antibodies. Of these, desmoglein 1 and desmoglein 3 are the best characterized. Desmoglein 1 is a 160-kDa molecule that is expressed in greater intensity in the upper layers of the epidermis but in only small quantities in squamous mucosa. It is the target antigen in pemphigus foliaceus. Desmoglein 3 is a 130-kDa molecule that localizes primarily in the deeper layer of epidermis and throughout the thickness of squamous mucosae. It is the target antigen in pemphigus vulgaris (36). Since mucosal epithelium expresses mainly desmoglein 3 but skin expresses both desmoglein 1 and 3, damage by antibodies to desmoglein 3 alone results in oral lesions with or without skin lesions. If both desmoglein 3 and desmoglein 1 antibodies are present, cutaneous lesions, as well as mucosal lesions, appear, and the disease tends to be more severe (37).
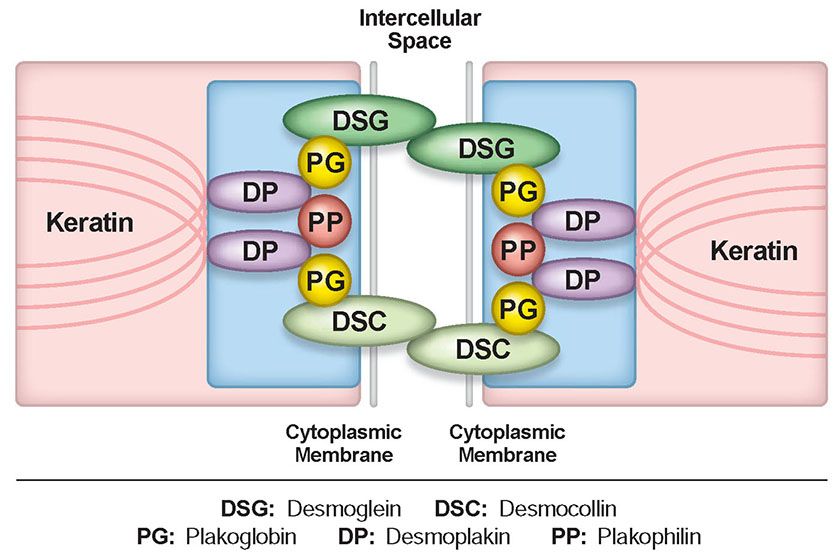
Figure 9-8 The desmosome. The desmosome complex includes desmogleins and desmocollins as transmembrane components, and plakoglobin, plakophilin, and desmoplakin as cytoplasmic components. The cytoplasmic domains of desmoglein and desmocollin associate with plakoglobin, which links intermediate filaments (keratin) to the desmosome through desmoplakin.
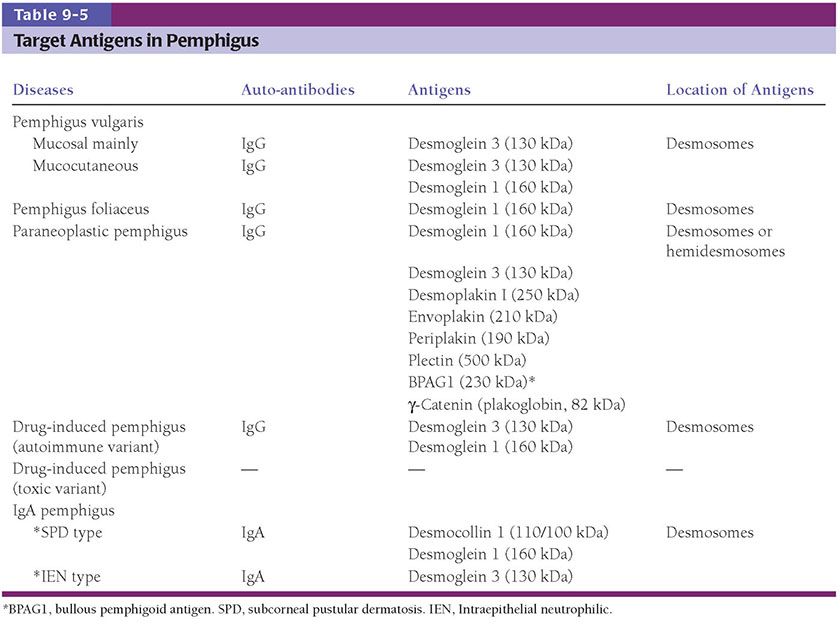
In summary, the localization and degree of expression of desmogleins 1 and 3 along with the presence of anti-desmoglein 1 antibodies alone, anti-desmoglein 3 antibodies alone, or both antibodies together reliably predict the mucosal and/or cutaneous involvement and the severity of pemphigus in each patient. Less is known about the desmocollins. Some cases of IgA pemphigus have had autoantibodies to desmocollin 1.
Pemphigus Vulgaris
Clinical Summary. This condition develops primarily in older individuals, presenting with large and flaccid bullae. They break easily and leave denuded areas that tend to increase in size by progressive peripheral detachment of the epidermis, leading in some cases to widespread cutaneous involvement. The lesions characteristically involve the oral mucosa, scalp, midface, sternum, groin, and pressure points. Oral lesions (Fig. 9.9A) are almost invariably present and may be the first manifestation of the disease (10% of cases) (38). Before corticosteroids became available, the mortality of this disease was high because of fluid loss and superinfection.
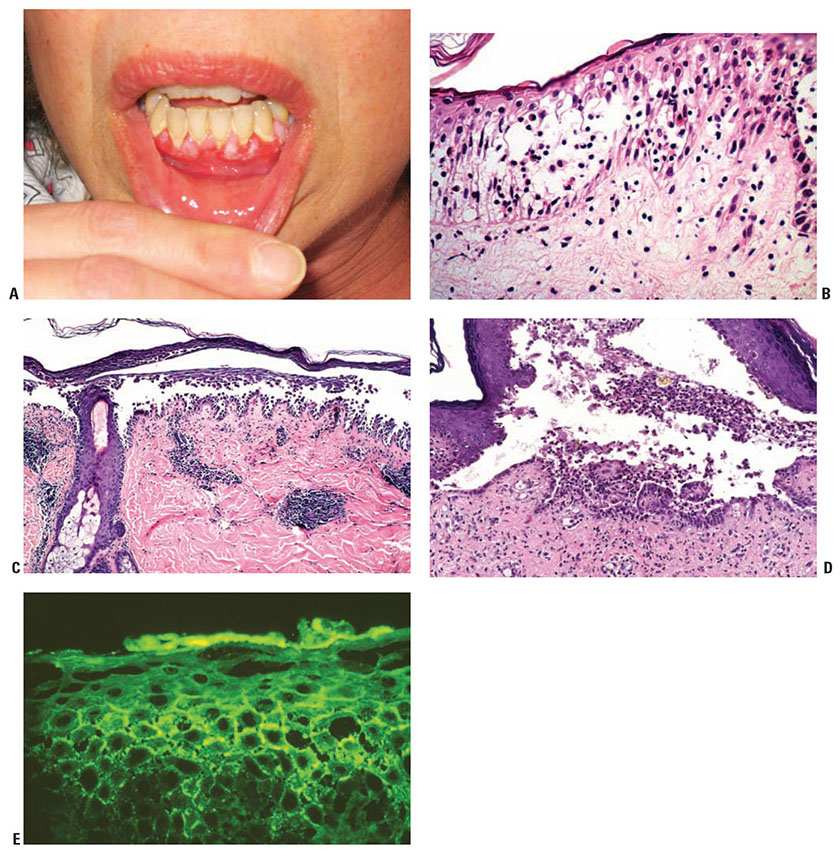
Figure 9-9 Pemphigus vulgaris. A: Multiple gingival erosions. (Courtesy of Kenneth Tsai, MD, PhD.) B: The earliest changes consist of intercellular edema with eosinophilic spongiosis leading to loss of intercellular bridges in the lower epidermis. C: An intraepidermal acantholytic blister has a suprabasal cleavage plane. Acantholysis may extend into adnexal structures and stratum spinosum. D: The suprabasal blister contains acantholytic cells, neutrophils, and eosinophils. Note the dermal papillae lined by a single layer of basal keratinocytes, so-called villi. E: There is lacelike squamous intercellular space deposition of IgG in the lower epidermis (direct immunofluorescence).
Histopathology. It is important that early blisters, preferably small ones, are selected for biopsy. Care should be given to keeping the epidermis attached to the dermis, because the torque applied in punch biopsies may separate the blister roof from the blister base. Therefore, one may use a refrigerant spray before excising the blister with a punch biopsy. A shave biopsy is a good method of obtaining an appropriate sample if delicately performed and the specimen is handled with care. If no recent blister is available, an old one may be moved into the neighboring skin by gentle vertical pressure with a finger (positive Nikolsky sign). The newly created cleavage will reveal early and specific histologic changes.
The earliest recognized change may be either eosinophilic spongiosis, or more commonly, “spongiosis” in the lower epidermis (Fig. 9-9B). This “spongiosis” may actually represent the earliest manifestation of acantholysis rather than true spongiosis as defined earlier. Acantholysis leads first to the formation of clefts, and then to blisters in a predominantly suprabasal location (Fig. 9-9C, D). The intraepithelial acantholysis extends into adnexal structures, or occasionally be higher in the stratum spinosum. The basal keratinocytes, although separated from one another through the loss of attachment, remain firmly attached to the dermis like a “row of tombstones.” Within the blister cavity, the acantholytic keratinocytes, singularly or in clusters, have rounded condensed cytoplasm about an enlarged nucleus with peripherally palisaded chromatin and enlarged nucleoli. In some patients, there are varying quantities of anti-desmoglein 1 and anti-desmoglein 3 antibodies, leading to variable planes of acantholysis. There is little inflammation in the early phase of blister formation. If present, it is usually a sparse, lymphocytic perivascular infiltrate accompanied by dermal edema. If, however, eosinophilic spongiosis is apparent, numerous eosinophils may infiltrate the dermis.
The phenomenon of eosinophilic spongiosis occurs occasionally in other blistering diseases, particularly in their early phases, including contact dermatitis, pemphigus foliaceus, bullous pemphigoid, herpes gestationis, drug eruptions, spongiotic arthropod bite reactions, and transient acantholytic dermatosis. Several important changes ensue as the lesions age. First, a mixed inflammatory cell reaction consisting of neutrophils, lymphocytes, macrophages, and eosinophils may develop. Because of the instability of the blister roof, erosion and ulceration may occur. Older blisters may also have several layers of keratinocytes at the blister base because of keratinocyte migration and proliferation. Lastly, there may be considerable downward growth of epidermal strands, giving rise to so-called villi (Fig. 9-9D).
The evaluation of patients with only oral lesions is difficult, because intact blisters are rarely encountered due to the trauma of mastication, and biopsies may show only erosion and ulceration. Indeed, it is best to sample the edge of a denuded area with intact mucosa in an attempt to demonstrate the typical pathologic changes. Clinicians frequently cannot distinguish between an ulcer and the intact mucosa, as both are often white and shaggy. In patients with only oral lesions, biopsies of intact oral mucosa for DIF testing are more sensitive than biopsies of lesions for routine light microscopic evaluation. Therefore, biopsy from the maxillary and upper buccal mucosa is necessary when there is extensive ulceration. Serum contents may insudate into the squamous intercellular substance.
Cytologic examination using a Tzanck preparation is useful for the rapid demonstration of acantholytic epidermal keratinocytes in the blisters of pemphigus vulgaris. For this purpose, a smear is taken from the underside of the roof and from the base of an early freshly opened bulla. Giemsa stain is applied with subsequent rinsing and air-drying. Because acantholytic keratinocytes are occasionally seen in various nonacantholytic vesiculobullous or pustular diseases as a result of secondary acantholysis, cytologic examination represents merely a preliminary test and should not supplant histologic examination. The acantholytic keratinocytes often mimic herpetically infected cells.
Immunofluorescence (IF) Testing. The edge of a blister with intact surrounding normal skin, uninvolved skin adjacent to a blister or adjacent erythematous skin should be supplied for study. The tissue may be snap-frozen or transported in Michel medium. DIF testing is a very reliable and sensitive diagnostic test for pemphigus vulgaris, in that it demonstrates lacelike IgG in the squamous intercellular/cell surface areas in up to 95% of cases, including early cases and those with very few lesions, and in up to 100% of cases with active disease (Fig. 9-9E). It remains positive, after clinical disease has subsided. In late lesions, when acantholysis is well developed, the lacelike intercellular/cell surface pattern of IgG may become dot-like, paralleling electron microscopic findings and correlating with aggregation of desmosomes on the cell surface. Negative DIF findings when the patient is in remission may be a good prognostic indicator (39). At this time, DIF testing is incorrectly thought by most to be free of false positives; however, false positives may occur. On occasion, it may be difficult to distinguish intercellular staining of pemphigus from nonspecific staining; for example, epidermis in spongiotic dermatitis, psoriasis, bullous impetigo, and adjacent to ulcers secondary to a number of disorders may have insudation of serum into the intercellular substance. Often IgM, IgA, fibrinogen, and albumin are present as well, indicating nonspecific trapping in the false-positive tests. Immunoperoxidase methods have achieved roughly the same sensitivity as the IF method, but they have not replaced IF testing as the prime diagnostic tool.
For IIF testing, unfixed frozen sections of guinea pig esophagus, monkey esophagus, or normal human skin are used as substrate. In general, monkey esophagus is the best substrate for indirect IF tests. Circulating IgG autoantibody is demonstrated in the squamous intercellular substance in 80% to 90% of cases (40), and the titer correlates with disease activity. False-positive IIF tests occur. In a series of 1,500 patients with circulating pemphigus antibodies, approximately 1% had no evidence of clinical disease. Antibodies that mimic or that may give in vitro deposition in stratified squamous epithelium in the absence of pemphigus have been reported in burns, penicillin allergy, toxic epidermal necrolysis (TEN), systemic lupus erythematosus (SLE), myasthenia gravis, bullous pemphigoid, CP, lichen planus, and in patients with antibodies directed against blood groups A and B. Such antibodies are present in low titer and are thought to be nonpathogenic. Anti-desmoglein autoantibodies are sometimes found in patients with no bullous disease. For example, they have been found in patients with silicosis (41) and in relatives of patients with pemphigus vulgaris (42).
Pathogenesis. Compelling evidence has accumulated that IgG autoantibodies against desmoglein 3 and desmoglein 1 are pathogenic and play a primary role in inducing the blister formation in pemphigus. Affinity-purified IgG from pemphigus vulgaris sera recognize the extracellular domain of desmoglein 3 and cause suprabasal acantholysis when injected into neonatal mice (43). Furthermore, when anti-desmoglein 3 IgG from pemphigus vulgaris is immunoabsorbed with the extracellular domains of desmoglein 3, those sera no longer have the ability to cause blisters in neonatal mice (44). Although the pathogenic role of anti-desmoglein autoantibodies in blister formation is assured, the exact sequence of events that occur after antibody binding is not totally understood. Conformational epitope mapping of desmoglein 3 in pemphigus vulgaris suggest that the amino terminal residue 1-161 is the target of autoantibodies (45). This segment is in the extracellular domain that is essential for cell–cell adhesion. One possibility is that these antibodies interfere directly with the adhesive interaction of desmogleins between cells by steric hindrance. Another possibility is that the disruption of cell–cell adhesion is mediated by signal transduction. Proteinases, likely induced by pemphigus antigen–antibody union, are thought to play an important role in acantholysis. Although complement fixation by pemphigus antibodies may promote acantholysis, acantholysis occurs in experimental systems in the absence of complement. The stimulus for the formation of autoantibodies is unknown, although drugs, viral infection, trauma, ionizing radiation, and PUVA therapy have been implicated because they have preceded the onset of pemphigus. Pemphigus vulgaris is rarely associated with internal cancer, Castleman disease, thymoma, myasthenia gravis, localized scleroderma, Graves disease, and SLE.
The involvement of the oral mucosa in pemphigus vulgaris is due to the expression of desmoglein 3 and the much lower expression of desmoglein 1 in the squamous mucosa.
Ultrastructural Study. The intercellular cement substance, or glycocalyx, is partially or entirely lysed in lesions with early acantholysis. There is widening of the intercellular spaces with intact desmosomes. As the intercellular space is widened, there is separation of the two opposing attachment plaques of the desmosomes, so that single attachment plaques, with adherent tonofilaments, are seen at the periphery of keratinocytes. As acantholysis progresses, the desmosomes gradually disappear and the keratinocytes develop numerous cytoplasmic processes that often interdigitate with one another. All of the early ultrastructural changes in pemphigus vulgaris are extracellular. Only subsequent to the dissolution of the desmosomes does retraction of the tonofilaments to the perinuclear area develop with ultimate degeneration of the acantholytic cells. The cohesion of the basal keratinocytes with the basement membrane zone is not affected in pemphigus vulgaris because of the preservation of structures connecting the basal keratinocytes with the dermis. Immunoelectron microscopy shows that the immunoglobulins are deposited on the surface of the keratinocytes in a discontinuous globular pattern in the extracellular domains of desmosomes (46).
Differential Diagnosis. In early blisters that are free of secondary changes, such as the degeneration or regeneration of epidermal cells, the histopathology of pemphigus vulgaris is characteristic. Important differential diagnoses include Hailey–Hailey disease and transient acantholytic dermatosis. Hailey–Hailey disease has full-thickness (“dilapidated brick wall”) acantholysis, epidermal hyperplasia, and an impetiginized scale crust. The acantholysis does not extend down follicles as it does in pemphigus. Transient acantholytic dermatosis may exhibit small foci of intraepidermal acantholysis, but these are only a few rete wide in contrast to the uniform widespread acantholysis observed in biopsies of pemphigus vulgaris. Disorders that are characterized by focal acantholytic dyskeratosis are readily separated from pemphigus vulgaris by the presence of abnormal granular keratinocytes and parakeratotic cells, so-called corps ronds and corps grains. Although light microscopic examination of pemphigus lesions is important, positive DIF is the gold standard in diagnosis at this time and must be pursued in all cases in which pemphigus vulgaris is considered.
Principles of Management. Management of pemphigus vulgaris may be challenging as patients are at risk of bacterial colonization of open wounds and secondary infection with both bacteria and viruses. Most patients require immunosuppression, often starting with systemic corticosteroids and transitioning to a steroid-sparing immunosuppressive agent, such as mycophenolate or azathioprine. Rituximab is a promising agent that may offer more targeted treatment, and has shown impressive results in limited studies to date.
Pemphigus Vegetans
Clinical Summary. This is an uncommon variant of pemphigus vulgaris, comprising only 1% to 2% of cases (41). Historically, pemphigus vegetans has been divided into the Neumann type and Hallopeau type. In the more severe Neumann type, the disease begins and ends as pemphigus vulgaris, but many of the denuded areas heal with verrucous vegetations that may contain small pustules in early stages. The Hallopeau type is relatively benign, having pustules as the primary lesions instead of bullae. Their development is followed by the formation of gradually enlarging verrucous vegetations, especially in intertriginous areas.
Histopathology. In the Neumann type, the early lesions consist of bullae and denuded areas that have the same histologic picture as that of pemphigus vulgaris. As the lesions age, however, there is formation of villi and verrucous epidermal hyperplasia. Numerous eosinophils are present within the epidermis and dermis, producing both eosinophilic spongiosis and eosinophilic pustules (Fig. 9-10A). Acantholysis may not be present in older lesions. In the Hallopeau type, the early lesions consist of pustules arising on normal skin with acantholysis and formation of small clefts, many in a suprabasal location. The clefts are filled with numerous eosinophils and degenerated acantholytic epidermal cells (Fig. 9-10B). Early lesions may reveal more eosinophilic abscesses than in the Neumann type. The subsequent verrucous lesions are histologically identical to the Neumann type.
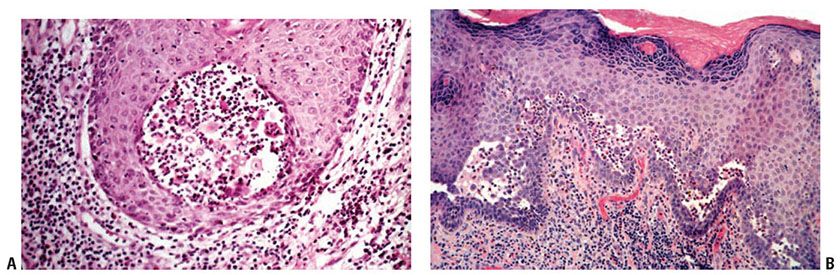
Figure 9-10 Pemphigus vegetans. A: Intraepidermal abscesses are composed of eosinophils and a few acantholytic keratinocytes. B: There is marked acanthosis and suprabasal clefts containing eosinophils.
IF Testing. DIF examination reveals squamous intercellular IgG in all reported cases.
Pathogenesis. Pemphigus vegetans is a variant of pemphigus vulgaris in which verrucous vegetations develop. It is unclear why such vegetations develop in some cases of pemphigus vulgaris and not in others. However, they tend to develop in areas of relative occlusion and maceration with subsequent bacterial infection, suggesting a response to superinfection.
Differential Diagnosis. The principal differential diagnosis is pyoderma vegetans, a condition often associated with inflammatory bowel disease, particularly ulcerative colitis. It can mimic pemphigus vegetans clinically and histologically (47). In pyoderma vegetans, neutrophils are more commonly found, and intraepidermal eosinophilic abscesses and acantholysis are rare. DIF is negative in pyoderma vegetans (48). Halogenoderma and blastomycosis-like pyoderma must be excluded as well (Chapters 11 and 23).
Principles of Management. Treatment is similar to that of pemphigus vulgaris, though local care with appropriate topical antimicrobials and in some cases anti-inflammatory therapies may be beneficial.
Pemphigus Foliaceus
Clinical Summary. Usually developing in middle-aged individuals, pemphigus foliaceus may have a chronic generalized course or may rarely present as an exfoliative dermatitis. Patients may present with flaccid bullae that usually arise on an erythematous base or as scaly, crusted erosions without evident blisters (Fig. 9-11A). Because of their superficial location, the blisters break easily, leaving shallow erosions rather than the denuded areas seen in pemphigus vulgaris. Oral lesions do not occur. The Nikolsky sign is positive, and Tzanck preparation reveals acantholytic granular keratinocytes. Fogo selvagem (endemic pemphigus foliaceus) is clinically, histologically, and immunologically indistinguishable from pemphigus foliaceus (48). It develops in those who live or visit areas close to rivers and streams in Brazil, the peak incidence being at the end of the rainy season. The cause of fogo selvagem is unknown, but substantial epidemiologic evidence suggests that it is precipitated by an environmental factor. A case control study found that farmers exposed to black fly (Simulium pruinosum) bites were much more likely to develop fogo selvagem than farmers who were not bitten (49). Sunlight may exacerbate the condition (50).
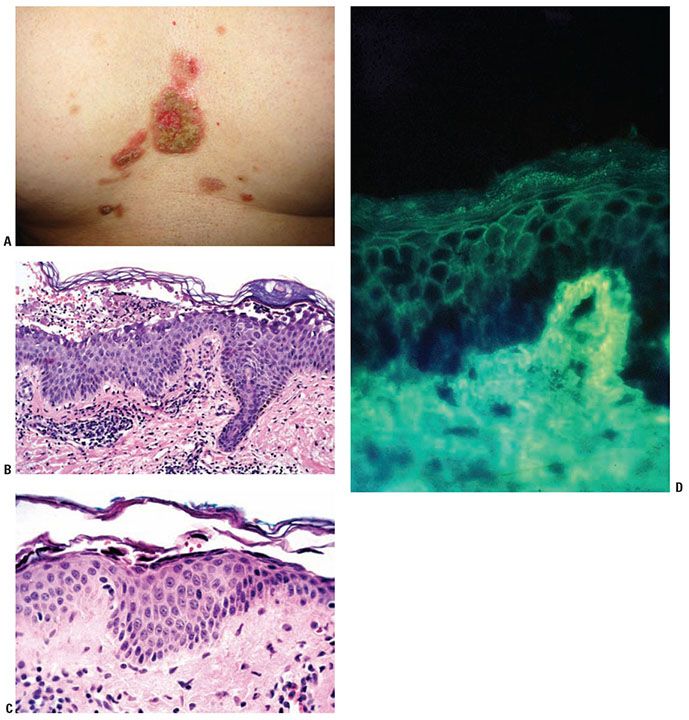
Figure 9-11 Pemphigus foliaceus. A: Erosions with thick crusts on the central chest. (Courtesy of Peter Lio, MD.) B: A subcorneal blister with acantholytic cells and neutrophils in the cavity. C: In the setting of a subcorneal blister, these dyskeratotic acantholytic granular cells are diagnostic of pemphigus foliaceus. D: The intercellular IgG is deposited in the intercellular spaces throughout the epidermis. Rarely, as in this case, the IgG is present in greater quantity in the superficial layers. In this biopsy, the roof of the blister is eroded, a frequent event, leaving a diminished but acantholytic granular layer.
Histopathology. The earliest change consists of acantholysis in the upper epidermis, within or adjacent to the granular layer, leading to a subcorneal bulla in some instances (Fig. 9-11B, C). More commonly, enlargement of the cleft leads to detachment of the stratum corneum without bulla being seen. The acantholytic granular cells may be spindled and not rounded because their cell shape is fixed at the time of IgG binding to the desmoglein or that the binding to the desmoglein permits differentiation to a granular keratinocyte architecture. The number of acantholytic keratinocytes is usually small, often requiring a careful search to identify them. Secondary clefts may develop, leading to detachment of the epidermis in its mid-level. These clefts may extend to above the basal layer, rarely giving rise to limited areas of suprabasal separation. In the setting of a subcorneal acantholytic blister, dyskeratotic granular keratinocytes are diagnostic for this disorder. Eosinophilic spongiosis may be prominent with intraepidermal eosinophilic pustules. Thus, the histologic features of pemphigus foliaceus may have three patterns diagnostic for this disorder: (a) eosinophilic spongiosis;(b) a subcorneal blister, often with few acantholytic keratinocytes; and (c) a subcorneal blister with dyskeratotic granular keratinocytes (Fig. 9-11C). The character of the inflammatory infiltrate is variable and depends on the age of the lesion, whether a blister is present, whether the superficial portion of the epidermis has been detached, and whether there is impetiginization or necrosis of the blister roof.
IF Testing. DIF testing of perilesional skin is positive in most of the cases. Two patterns of pemphigus antibody deposition have been described. In most cases, there is full-thickness squamous intercellular substance deposition of IgG. Rarely, IgG may be localized only to the superficial portion of the epidermis (Fig. 9-11D). IIF testing of serum reveals squamous intercellular substance deposition of IgG in 80% to 90% of cases.
Pathogenesis. As in pemphigus vulgaris, the autoantibodies of pemphigus foliaceus are pathogenic. During the course of the disease, the antibody levels fluctuate, and have some correlation with disease activity. The pemphigus foliaceus antigen, desmoglein 1, is expressed more intensely in the upper layers of the epidermis (35,36), which explains the superficial cleavage plane of pemphigus foliaceus. Interestingly, in staphylococcal scalded skin syndrome (SSSS), and its localized form, bullous impetigo, the exfoliative exotoxin produced by S. aureus specifically binds and cleaves desmoglein 1, resulting in blister formation at identical levels of the epidermis as pemphigus foliaceus (51,52). In addition, desmoglein 1 is concentrated in the upper torso and is less prominent in the buccal mucosa, scalp, and lower torso, correlating with lesion distribution (53).
Ultrastructural Study. There is early loss of intercellular cement substance within the lower epidermis associated with a decrease in the number of desmosomes and formation of tortuous microvilli from the keratinocyte surface. However, acantholysis is most pronounced in the upper layers of the epidermis. In the mid-epidermis, many keratinocytes have perinuclear arrangement of the tonofilaments and homogenization of the perinuclear tonofilament bundles as evidence of dysmaturation.
Differential Diagnosis. The differential diagnosis includes SSSS, impetigo, subcorneal pustular dermatosis (Sneddon–Wilkinson), and IgA pemphigus (see differential diagnosis of IgA pemphigus). IF testing may be required to separate SSSS from pemphigus foliaceus, since a small number of acantholytic cells may be observed in SSSS. The lesions of pemphigus foliaceus may become impetiginized and secondarily altered, producing pustules as in impetigo, subcorneal pustular dermatosis, and IgA pemphigus. Pemphigus foliaceus contains more acantholytic keratinocytes than do the other two disorders, and pustules are the primary lesions in subcorneal pustular dermatosis. Because the lesions of pemphigus foliaceus may become superinfected, the finding of bacteria does not confirm a diagnosis of bullous impetigo; therefore, IF testing is critical. Subcorneal pustular dermatosis usually produces large dome-shaped subcorneal pustules rather than the flaccid flat blisters and pustules of pemphigus foliaceus.
Principles of Management. Pemphigus foliaceus, like pemphigus vulgaris, often requires systemic immunosuppressive therapies with corticosteroids and a transition to a nonsteroid immunosuppressive agent. Patients are also at risk of viral or bacterial superinfection of open areas, particularly when immunosuppressed.
Pemphigus Erythematosus
Clinical Summary. Also known as Senear–Usher syndrome, pemphigus erythematosus is a variant of pemphigus foliaceus that gets its name from its lupus erythematosus–like clinical appearance, in which the erythematous plaques and patches are in a butterfly distribution. They may remain localized to this area or may become generalized. No oral lesions are observed.
Histopathology. The light microscopic features are identical to those of pemphigus foliaceus in the vast majority of cases (Fig. 9-12). Interface dermatitis has been apparent in rare cases, making distinction from lupus erythematosus difficult.
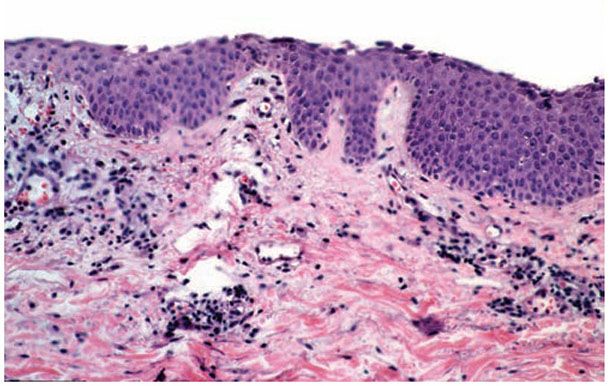
Figure 9-12 Pemphigus erythematosus. The histological appearances are identical to those of pemphigus foliaceus. In this biopsy, the roof of the vesicle is eroded, which is a frequent event, leaving a diminished but acantholytic granular layer.
IF Testing. DIF testing of perilesional skin reveals squamous intercellular substance deposition of IgG in greater than 75% of cases, and granular deposition of IgM and IgG (i.e., a positive lupus band test) at the dermoepidermal junction. IIF study using monkey esophagus as substrate reveals squamous intercellular substance deposition of IgG in 80% of cases. Antinuclear antibodies are observed in 30% to 80% of cases.
Ultrastructural Study. Pemphigus erythematosus is identical to pemphigus foliaceus in its epidermal ultrastructural alterations.
Differential Diagnosis. The differential diagnosis is the same as in pemphigus foliaceus. The presence of interface dermatitis in some cases leads to confusion with lupus erythematosus and PNP. Subcorneal acantholysis is not a feature of lupus erythematosus.
Principles of Management. Management of this rare form of pemphigus may be challenging as some patients may have features of SLE as well; immunosuppressive therapy is the mainstay.
Pemphigus Herpetiformis
Clinical Summary. Pemphigus herpetiformis combines the clinical features of dermatitis herpetiformis with the immunologic and histologic features of pemphigus, usually the foliaceus type (54). This variant deserves recognition only because of its clinical presentation. Patients present with pruritic, erythematous, vesicular, or papular lesions, often in herpetiform pattern. Mucous membranes are uncommonly involved.
Histopathology. There is eosinophilic spongiosis with or without acantholysis. Variable numbers of neutrophils may be present, leading to neutrophilic spongiosis or subcorneal pustules with both eosinophils and neutrophils.
IF Testing. Because the clinical presentation and histologic findings are often atypical, the most reliable basis for diagnosis of pemphigus herpetiformis is IF study. DIF of perilesional skin shows deposition of IgG on the surface of keratinocytes primarily in the upper epidermis (55). IIF has demonstrated circulating IgG anti-epithelial cell surface autoantibodies most directed against desmoglein 1, the pemphigus foliaceus antigen. A few cases with antibodies to the pemphigus vulgaris antigen, desmoglein 3, have been reported (55).
Principles of Management. Treatment is similar to pemphigus foliaceus, above.
Drug-Induced Pemphigus
Although immunologic features identical to those of idiopathic pemphigus are reported in most cases of drug-induced pemphigus (56), evidence indicates that some drugs may induce acantholysis without production of antibodies (57). The offending drugs, most frequently penicillamine, captopril, and penicillin derivatives, often contain sulfhydryl groups (58). The earliest clinical manifestation is a nonspecific morbilliform or urticarial eruption. In penicillamine-induced pemphigus, the eruption is characteristic and has been labeled “toxic pre-pemphigus rash (58).” Subsequently, the characteristic lesions of pemphigus develop. Upon cessation of drug therapy, the rash regresses in those patients who do NOT have pemphigus antibodies in contrast to those patients with pemphigus antibodies, in whom the rash frequently persists. This latter subset has the clinical waxing and waning course of pemphigus.
Histopathology. The findings in the early eruption are nonspecific, consisting of spongiosis, parakeratosis, and a variable dermal infiltrate. Well-developed lesions are essentially identical to those of pemphigus foliaceus or pemphigus vulgaris. Eosinophilic spongiosis may be prominent.
IF Testing. DIF testing is positive in approximately 90% of patients with drug-induced pemphigus (58). IIF study of serum reveals circulating squamous intercellular substance IgG antibodies in 70% of cases. The antibody titers are usually low and do not appear to correlate with the severity of disease.
Pathogenesis.
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


