Microtia reconstruction is a challenging endeavor that has seen significant technique evolution. It is important to educate patients and their families to determine the best hearing rehabilitation and ear reconstructive options. Microtia is often associated with aural atresia, hearing loss, and craniofacial syndromes. Optimal care is provided by multiple disciplines, including a reconstructive surgeon, an otologic surgeon, an audiologist, and a craniofacial pediatrician. Microtia management includes observation, prosthetic ear, autologous cartilage reconstruction, or alloplastic implant placement. Hearing management options are observation, bone conduction sound processor, or atresiaplasty with and without hearing aids. Appropriate counseling should be done to manage expectations.
Key points
- •
Children with outer ear anomalies should have diagnostic audiological assessment.
- •
Management of hearing should be considered when developing a plan for auricular reconstruction because it may impact the timing and order of procedure(s) because atresia is present in 75% of microtia.
- •
Options for management of microtia include observation, prosthetic management, and reconstruction.
- •
Reconstruction options include staged autologous costal cartilage reconstruction and single-stage reconstruction with alloplastic framework.
- •
Families should be educated on all treatment options in terms of both hearing rehabilitation and reconstruction options.
Introduction
Microtia, or small or malformed ear, occurs with an incidence of 1 to 10 per 10,000 births. Although associated with many syndromes, it occurs in isolation and unilaterally in most cases. The right side is more commonly affected, and boys have a 30% higher affected rate than girls. Ethnic groups with the highest incidence include Andeans, Native Americans, Asians, and Hispanics. Aural atresia is found with microtia in 75% of cases.
Embryologically, the external ear begins to form at 6 weeks from tissue derived from first and second branchial arches. The 6 hillocks of His become the tragus, helix, concha cymba, antihelix, and antitragus. A meatal plug expands and forms the tympanic membrane by 13 weeks. At 18 weeks, the meatus is fully formed, as are all parts of the external ear.
In addition to ethnicity and male sex, risk factors for microtia include low birth weight and acute maternal illness. In utero exposure to teratogens such has thalidomide and retinoids are strongly associated with microtia. Higher levels of folate ingestion during pregnancy have been found to reduce the incidence of microtia. The precise mechanism for the development of microtia is a topic of ongoing research.
The microtia phenotype appears in a spectrum of disorders of which the most common include craniofacial microsomia, Goldenhar, and Treacher Collins. Multiple other syndromes or genetic causes have been identified and are associated with microtia in less than 50% of cases ( Box 1 ). Although the contralateral ear appears normal size in most cases, detailed measurement reveals that it is actually smaller than a normal control group. There may be other abnormalities that have not fully been evaluated or discovered in their association with microtia. For example, a recent study found high correlations with chest wall deformities when detailed analysis of thoracic imaging was performed.
Auriculo-condylar
Bixler (hypertelorism-microtia-clefting)
Bosley-Salih-Alorainy
Branchio-oculo-facial
Branchio-oto-renal/branchio-otic
CHARGE
Fraser
Kabuki
Klippel-Feil
Labyrinthine aplasia
Meier-Gorlin
Miller
Nager
Oculo-auricular
Pallister-Hall
Townes-Brocks
Treacher Collins
Wildervanck (cervico-oculo-acoustic)
Introduction
Microtia, or small or malformed ear, occurs with an incidence of 1 to 10 per 10,000 births. Although associated with many syndromes, it occurs in isolation and unilaterally in most cases. The right side is more commonly affected, and boys have a 30% higher affected rate than girls. Ethnic groups with the highest incidence include Andeans, Native Americans, Asians, and Hispanics. Aural atresia is found with microtia in 75% of cases.
Embryologically, the external ear begins to form at 6 weeks from tissue derived from first and second branchial arches. The 6 hillocks of His become the tragus, helix, concha cymba, antihelix, and antitragus. A meatal plug expands and forms the tympanic membrane by 13 weeks. At 18 weeks, the meatus is fully formed, as are all parts of the external ear.
In addition to ethnicity and male sex, risk factors for microtia include low birth weight and acute maternal illness. In utero exposure to teratogens such has thalidomide and retinoids are strongly associated with microtia. Higher levels of folate ingestion during pregnancy have been found to reduce the incidence of microtia. The precise mechanism for the development of microtia is a topic of ongoing research.
The microtia phenotype appears in a spectrum of disorders of which the most common include craniofacial microsomia, Goldenhar, and Treacher Collins. Multiple other syndromes or genetic causes have been identified and are associated with microtia in less than 50% of cases ( Box 1 ). Although the contralateral ear appears normal size in most cases, detailed measurement reveals that it is actually smaller than a normal control group. There may be other abnormalities that have not fully been evaluated or discovered in their association with microtia. For example, a recent study found high correlations with chest wall deformities when detailed analysis of thoracic imaging was performed.
Auriculo-condylar
Bixler (hypertelorism-microtia-clefting)
Bosley-Salih-Alorainy
Branchio-oculo-facial
Branchio-oto-renal/branchio-otic
CHARGE
Fraser
Kabuki
Klippel-Feil
Labyrinthine aplasia
Meier-Gorlin
Miller
Nager
Oculo-auricular
Pallister-Hall
Townes-Brocks
Treacher Collins
Wildervanck (cervico-oculo-acoustic)
Diagnosis and evaluation
Patients with microtia and malformed ears are diagnosed at birth and should undergo audiological testing. Microtic ears should be carefully examined for the presence of an ear canal. Newborn hearing screen should be performed in all ears with a patent ear canal. Over the long term, even if unilateral microtia is present, an otolaryngologist and audiologist should maintain regular clinical encounters because the contralateral ear is at higher risk for abnormalities than the general population.
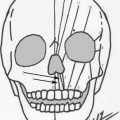
If bilateral atresia is present, diagnostic brainstem auditory-evoked responses should be performed. Amplification and enrollment in early intervention should be initiated, ideally within the first few months of life. On examination, it is important to accurately describe the malformed ear. Fig. 1 shows normal external auricle anatomy. Multiple classification schemes are used to describe the degree of microtia. Fig. 2 displays 4 classes of microtia as proposed by Marx and modified by Rogers. In grade I, the ear is small or abnormal, but all landmarks are discernible. In grade II, some of the landmarks are identifiable. Grade III has very small external auricle components, often only a skin tag. Grade IV is anotia. Nagata proposed a classification scheme of descriptive terms, including lobule-type, concha-type, and small concha-type. In lobule type, there is a remnant of the lobule and auricle with no canal, concha, nor tragus. Concha-type has variable presence of the lobule and tragus. Small concha-type has a small indentation of the concha and remnant of lobule and auricle. He also used anotia and atypical in the scheme, in which the ear did not fit into the above types.
Other classification schemes to summarize the craniofacial anomalies associated with microtia have been described. OMENS is a classification system for hemifacial microsomia proposed by Mulliken that examines variables of orbital, mandibular, ear, neural, and soft tissue phenotypes. Research using OMENS has found that 67% of patients with hemifacial microsomia have extracraniofacial anomalies and 26% have cardiac anomalies, which has modified the classification scheme to OMENS-Plus if an extracraniofacial anomaly is present. Recently, a classification scheme called HEAR MAPS that incorporates multiple other staging systems was proposed to improve communication among the multiple disciplines of providers ( Table 1 ).
| Hear | Air-bone gap (dB HL) |
| Ear | Microtia grade 1–4 |
| Atresia | Jahrsdoerfer CT scale (1–10) |
| Remnant earlobe | Grade 1–4 |
| Mandible asymmetry | Grade 1–4 |
| Asymmetry soft tissue | Grade 1–4 |
| Paresis of the facial nerve | House-Brackmann scale (1–6) |
| Syndrome | (Yes/No) |
Indications and counseling
Indications for microtia management should be based on a discussion with the patient and patient’s family. There is an overall increased rate of depression and anxiety in microtia patients compared with a control cohort. Studies have shown reduced psychological stressors after undergoing microtia reconstruction and overall patient satisfaction. Furthermore, a reconstructed auricle will permit retention of a hearing aid or glasses.
Most microtia patients also have aural atresia, and the management of the conductive hearing loss has implications on microtia reconstruction. All options, both for hearing rehabilitation and for auricular reconstruction, should be thoroughly discussed with the patient and family. It is important for the surgeon and the family to generate a cohesive plan that includes management of the ear and hearing. The hearing management options are summarized in Table 2 , and the microtia management options are summarized in Table 3 .
| Approach | Advantages | Disadvantages |
|---|---|---|
| Observation | Minimize risk | Unilateral hearing loss |
| Band-retained bone conduction sound processor | No surgery | Cosmesis Device required Comfort |
| Osseointegrated implant-retained bone conduction sound processor | Simple surgery Predictable Excellent hearing result Magnetic option available | Cosmesis Device required Must be at least 5 years old Soft tissue issues (nonmagnetic) |
| Atresiaplasty | Cosmesis Accommodation of ear level hearing aid if necessary No device | Complex surgery Less predictable result Modest hearing benefit Ongoing care required |
| Type | Details | Advantages | Disadvantages |
|---|---|---|---|
| Observation | No risk | Cosmesis Psychosocial issues | |
| Prosthetic | Adhesive retained | Appearance | Insecure Ongoing prosthetic care Daily maintenance Use restrictions |
| Implant retained | Appearance Secure retention | Multiple procedures Removal of remnant and soft tissue Ongoing prosthetic care Daily maintenance Use restrictions | |
| Reconstruction | Costal cartilage (autologous) | Autologous tissue Minimal maintenance Becomes sensate Atresia repair | Appearance Donor sites Multiple surgeries |
| Alloplastic | Less donor site morbidity Less variability in carving Appearance Single surgery | Foreign body More challenging to do atresia repair |
From a surgical planning perspective, one of the main decision points is atresiaplasty candidacy, which is based on high-resolution computed tomography (CT) of the temporal bones typically done around age 4 years. Obtaining the CT scan at about 4 years of age obviates sedation, allows for mastoid growth, and may mitigate the potential effects of radiation on the developing brain. This timing also allows for the CT scan to screen for occult congenital cholesteatoma. Traditionally, autologous costochondral microtia reconstruction was performed before atresiaplasty. More recently, multiple surgeons have reported atresiaplasty before or simultaneous with microtia reconstruction with good outcomes.
The only surgical reconstruction option for many years used autologous costochondral cartilage, and surgeons preferred to wait until the patient was at least 6 years of age for multiple reasons: (1) the contralateral ear is near full size, (2) the costochondral cartilage is of adequate size, and (3) the patient is able to understand the reconstructive options. The last point could be considered a disadvantage in that families may prefer to undergo the reconstruction as soon as possible and before school begins. The introduction of alloplastic reconstruction options has modified the timeline for decisions with the patient’s families because reconstruction as young as age 3 is now possible ( Fig. 3 ).