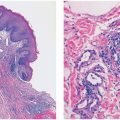
Figure 17-1 Primary systemic amyloidosis. A: Pinch purpura. Note the papules and ecchymotic appearance in the characteristic periocular location. B: Macroglossia. The patient’s tongue is enlarged with prominent indentations from continuous pressure against the teeth.
Histopathology. Examination of cutaneous lesions in the systemic form of the disease reveals faintly eosinophilic, amorphous, often fissured masses of amyloid deposited in the dermis and subcutaneous tissue. Accumulations of amyloid are frequently deposited close to the epidermis. They may or may not be separated from the overlying epidermis by a narrow zone of collagen (Fig. 17-2).
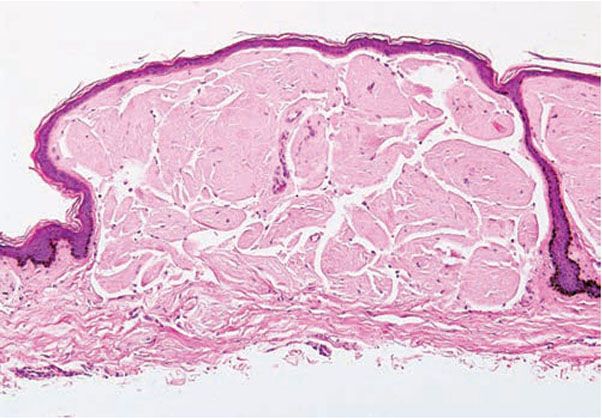
Figure 17-2 Primary systemic amyloidosis. There are amorphous, fissured masses of amyloid in the upper dermis. The amyloid material greatly resembles that observed in colloid milium (see Fig. 17-8, page 508).
They are rarely deposited around individual elastic fibers (17). The involvement of the walls of blood vessels is responsible for the frequent presence of extravasated erythrocytes. Inflammatory cells are lacking or scarce (14). Several mechanisms of bulla formation have been proposed: (a) the formation of clefts within large dermal amyloid deposits (18), (b) from disruption of basal keratinocytes and the basement membrane zone (14,18), or (c) secondary to inflammatory cell infiltration (18).
In the subcutaneous tissue, there may be large aggregates of amyloid with infiltration of the walls of blood vessels and so-called amyloid rings, which are formed by the deposition of amyloid around individual fat cells (19). The fat cells may then appear as if cemented together by the amyloid.
Even if there are no skin lesions, fine-needle aspirates of the abdominal fat often are of use in documenting the diagnosis, with reported sensitivities and specificities of 55% to 88% and 74% to 100%, respectively (20,21) (Fig. 17-3). Tissue biopsies of normal-appearing skin yield positive results in about 40% of all patients, showing small deposits in the walls of small blood vessels in the dermis or the subcutaneous tissue and occasionally also around eccrine glands and lipocytes. The forearm is the recommended area for biopsy (22).
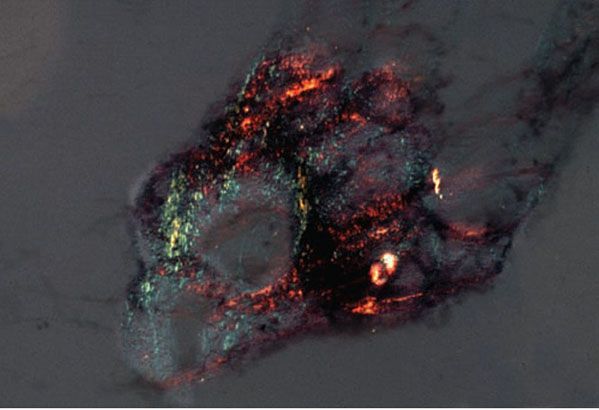
Figure 17-3 Primary systemic amyloidosis. Amyloid deposited in subcutaneous fat exhibits green birefringence under polarized light when stained with Congo red in this aspirate from the abdominal fat pad.
When localized to the skin, nodular amyloid deposits are surrounded by a dense plasmacytic infiltrate, and production of the immunoglobulin light chains is thought to occur locally.
Relationship with Multiple Myeloma. AL amyloidosis results from the production of monoclonal immunoglobulins and/or free light chains, usually Λ light chains, by an abnormal population of B cells. Up to 98% of the patients have detectable monoclonal protein in urine or serum (Bence-Jones protein) (23). If there is overtly malignant plasma cell neoplasia, it is referred to as multiple myeloma. Systemic amyloidosis is found in 5% to 15% of multiple myeloma patients. However, the majority of patients with AL amyloidosis do not have overt multiple myeloma (24). These patients have an underlying B-cell dyscrasia and, despite having no tumor masses, show an increased plasma cell population on bone marrow examination. It is uncertain whether or not these patients will inevitably develop multiple myeloma. Additionally, patients with nodular localized cutaneous amyloidosis (see below) may eventually develop systemic amyloidosis (25).
Pathogenesis. In the systemic form, the amyloid originates from monoclonal immunoglobulin light chains produced by plasma cells in the bone marrow. In the localized form, the amyloid is thought to be produced by the local plasma cell infiltrate, and as in the systemic form, the amyloid protein has immunocytochemical characteristics of AL amyloid (9,11).
Ultrastructurally, amyloid deposits consist of irregularly arranged, straight, nonbranching filaments that often appear hollow because their peripheries appear electron dense in comparison with their centers. These filaments are 6 to 7 nm in diameter but are of indeterminate length (26,27).
Principles of Management. Treatment modalities are aimed at eradicating the pathologic plasma cells and circulating free light chains and include immunosuppressants, chemotherapy, and peripheral blood stem cell transplantation. Patients require aggressive supportive care for affected organs and should be managed by an experienced multidisciplinary team. Irreversible end-organ damage may require transplantation.
AA (Serum Amyloid A) Amyloidosis
Clinical Summary. Also known as secondary systemic amyloidosis, AA amyloidosis can result from chronic inflammatory disease or recurring bouts of acute inflammation such as tuberculosis, complications of bronchiectasis, and chronic osteomyelitis. It is now relatively rare in industrialized countries since the development of modern antibiotic therapy, and most cases in the United States and Western Europe are associated with chronic rheumatoid arthritis or other inflammatory disorders (28). Rare cutaneous disease processes cited as causing secondary systemic amyloidosis include vasculitis and epidermolysis bullosa (28–31). At the time of diagnosis, most patients have renal insufficiency or the nephrotic syndrome (28). There are typically no cutaneous lesions, although rare reports of nodular and hemorrhagic bullous lesions of the skin containing AA amyloid have been reported (32,33).
Histopathology. AA amyloid is deposited in parenchymatous organs such as the kidneys, liver, spleen, and adrenals. These deposits are found first in the interstitium and blood vessel walls, then gradually replacing the parenchyma. Deposits within the glomeruli and peritubular tissues result in renal failure.
Fine-needle aspiration of subcutaneous fat, followed by Congo red staining, is the most sensitive method of diagnosis (20,34). Proteomic techniques can be utilized on unfixed adipose tissue from aspirates to reliably characterize AA protein (35). Tissue biopsy of skin with underlying subcutaneous fat demonstrates deposits of AA amyloid around lipocytes, in blood vessel walls, around eccrine glands, and sometimes free in the dermis (22).
Pathogenesis. AA amyloid is derived from serum amyloid A (SAA), an acute phase reactant, which is produced by the liver in response to inflammation and regulated by cytokines, including interleukin (IL)-1, IL-6, and tumor necrosis factor α. AA protein is then formed when SAA undergoes carboxy terminal cleavage (36,37). This process takes place within lysosomes of macrophages that are receiving antigenic stimulation by a variety of chronic diseases. The amyloid is then deposited extracellularly using heparan sulfate as a scaffold for fibril assembly (38).
Principles of Management. In AA amyloidosis, treatment is generally directed at controlling the underlying infection/inflammatory process.
Primary Localized Cutaneous Amyloidosis
Lichen Amyloidosis and Macular Amyloidosis
Clinical Summary. Lichen amyloidosis and macular amyloidosis are best considered as different manifestations of the same disease process, with lesions occurring due to local amyloid deposits limited to the skin, with the amyloid protein component derived from keratinocyte proteins (AK amyloid). Lichen amyloidosis is characterized by closely set, discrete, brown-red papules that often show some scaling and are most commonly located on the legs, especially the shins, although they may occur elsewhere (Fig. 17-4). These papules may coalesce to form plaques, often with verrucous surfaces sometimes resembling hypertrophic lichen planus or lichen simplex chronicus. Usually, the lesions of lichen amyloidosis itch severely. It is assumed by some authors that the pruritis leads to damage of keratinocytes by scratching and to subsequent production of amyloid (39,40).
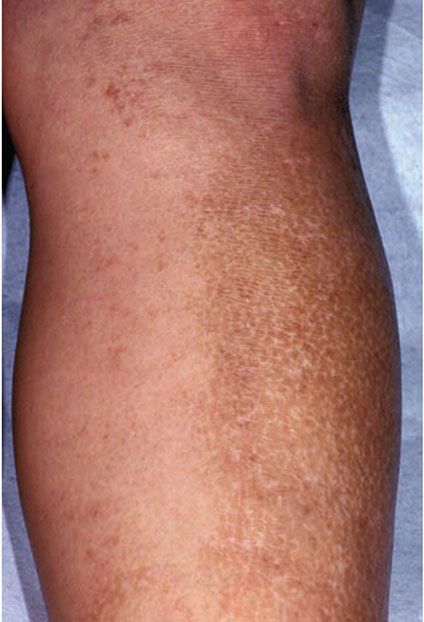
Figure 17-4 Lichen amyloidosis. There are thin, tan to brown papules coalescing into a larger plaque on the anterior lower leg.
Macular amyloidosis is characterized by pruritic macules showing pigmentation with a reticulated or rippled pattern (Fig. 17-5). Although macular amyloidosis may occur anywhere on the trunk or extremities, the upper back is a fairly common site (41). In Southeast Asia, where macular amyloidosis is common, prolonged friction from a rough nylon towel or a back scratcher is thought to be its cause (41). The eruption can be easily passed off as postinflammatory hyperpigmentation by physicians who are unfamiliar with the condition.
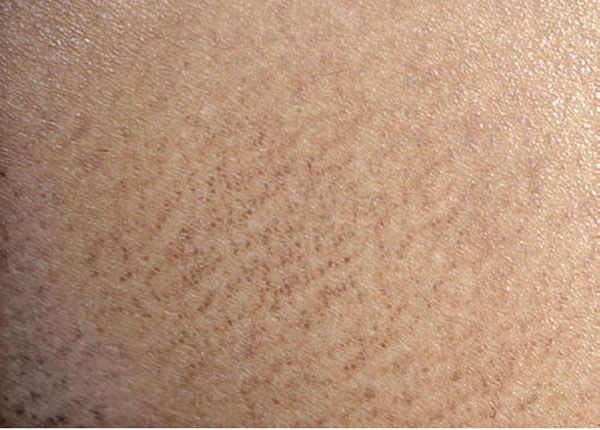
Figure 17-5 Macular amyloidosis. Note the rippled appearance of the hyperpigmentation in this close-up photograph of a patient’s central upper back, which is a characteristic location.
Primary cutaneous amyloidosis of the external ear is a rarely described variant of keratin-derived amyloidosis. In contrast to lichen and macular amyloidoses, it is generally not pruritic (42).
Macular amyloidosis and lichen amyloidosis sometimes occur together in the same patient (so-called biphasic amyloidosis), and lichen amyloidosis can arise in a setting of macular amyloidosis, presumably due to scratching (41,43). When treated by intralesional injection of steroids, the lichenoid lesions can become macular. Up to 10% of cases of macular and lichen amyloidoses are inherited in an autosomal dominant pattern. These cases of familial primary localized cutaneous amyloidosis can be mapped to a locus on 5p13.1-q11.2 that contains the OSMR gene, which encodes the IL-6 cytokine receptor OSMRβ (44).
Histopathology. Lichen and macular amyloidoses show deposits of amyloid that are limited to the papillary dermis. Although the deposits usually are smaller in macular amyloidosis than in lichen amyloidosis, differentiation of the two on the basis of the amount of amyloid is not possible. The two conditions actually differ only in the appearance of the epidermis, which is hyperplastic and hyperkeratotic in lichen amyloidosis (45). Occasionally, the amount of amyloid in macular amyloidosis is so small that it is missed even when special stains are used on frozen sections. In such instances, more than one biopsy or electron microscopy may be necessary to confirm the diagnosis (46).
In areas in which the entire dermal papilla is filled with amyloid, the amyloid appears homogeneous in both lichen and macular forms. In lesions in which the dermal papillae are only partially filled, as seen more often in macular amyloidosis, the amyloid has a globular appearance and resembles the colloid bodies found in lichen planus (Fig. 17-6). These amyloid bodies in some areas lie in direct contact with the overlying basal cells of the epidermis. Similar colloid bodies are also found in some sections within the epidermis, but in contrast with those located at the dermal–epidermal junction, they do not stain as amyloid. In addition, there often is a striking degree of pigmentary incontinence.

Figure 17-6 Lichen amyloidosis. There is hyperkeratosis and epidermal hyperplasia. Dermal papillae are rounded and contain globular deposits of amyloid. (A: H&E, original magnification ×40; B: H&E, original magnification ×100; C: H&E, original magnification ×200.)
Pathogenesis. The light microscopic findings in lichen and macular amyloidoses suggest that degenerating epidermal cells are discharged into the dermis, where they are converted into amyloid. This theory of primary localized cutaneous amyloid deposition, termed the fibrillar body theory, is supported by electron microscopy (47,48). Also on electron microscopy, the degenerating epidermal cells resemble the colloid bodies observed in lichen planus. They contain the following components: (a) tonofilaments; (b) degenerated, wavy tonofilaments that are thicker but less electron dense than normal tonofilaments; (c) lysosomes; and (d) typical filaments of amyloid, 6 to 10 nm thick, that are straight and nonbranching (48). It is postulated that the degenerated, wavy tonofilaments are recognized as foreign and are digested by the cell’s own lysosomes, producing amyloid filaments. A conversion of tonofilaments into amyloid filaments requires that the α-pleated sheet configuration of the tonofilaments change into the β configuration of amyloid (49). However, immunohistochemical studies suggest that components of the lamina densa and anchoring fibrils are also associated with amyloid deposits. Further ultrastructural examination shows disruption of the lamina densa overlying these deposits (50). As such, an alternative hypothesis of primary localized cutaneous amyloid deposition, the secretion theory, has been proposed. This theory suggests amyloid is secreted by disrupted basal cells and assembled at the dermal–epidermal junction. The amyloid then drops into the papillary dermis through a damaged lamina densa (51).
On direct immunofluorescence, all specimens of lichen or macular amyloidoses fluoresce positively for immunoglobulins or complement, particularly immunoglobulin M (IgM), complement C3, and immunoglobulin A (IgA) (52). κ and λ light chains are negative (53,54).
The epidermal derivation of the amyloid in lichen and macular amyloidoses is supported by histochemical and immunologic findings. In contrast to the amyloid of systemic amyloidosis, the amyloid of lichenoid and macular amyloidoses shows fluorescence for disulfide bonds as normally seen in the stratum corneum, suggesting that cross-linking of sulfhydryl groups occurs in amyloidogenesis (55). Furthermore, immunofluorescence and immunohistochemical studies have shown intense staining of the amyloid with antikeratin antibody (56,57). Also, strong cytokeratin 5 and 34βE12 immunopositivity in lichen and macular amyloidoses suggest the amyloid is derived primarily from basal keratinocytes (57). Other investigators have found direct amyloid fibril formation at the basal surfaces of living basal cells in lichen amyloidosis (58).
The amyloid that may be found in the stroma or in the adjacent connective tissue of basal cell carcinoma and other epithelial tumors has an appearance, on electron microscopy and direct immunofluorescence, similar to that of lichen and macular amyloidoses, suggesting that it too is derived from tonofilaments (59). This amyloid also shows positive staining with antikeratin antiserum similar to macular and lichen amyloidoses (57).
Principles of Management. Treatment is directed toward the relief of pruritis.
Nodular Localized Cutaneous Amyloidosis
Clinical Summary. Nodular localized cutaneous amyloidosis is a rare condition in which nodular deposits of AL amyloid are deposited in the skin but in which there is no apparent systemic involvement. One or several nodules are encountered, usually on the legs or face but occasionally elsewhere (8,9,60,61). The nodules may measure from a few millimeters to several centimeters in diameter and typically have a waxy tumefactive appearance. In their centers, the skin may appear atrophic with a white to yellow color (9). Anetodermic and bullous lesions may be encountered, and in exceptional instances, plaques are observed (60,62).
Histopathology. Beneath an atrophic epidermis are large masses of amyloid that extend through the entire dermis into the subcutaneous fat. Amyloid deposits are also found within the walls of blood vessels, in the membrana propria of the sweat glands, and around fat cells (8,25). A lymphoplasmacytic infiltrate is scattered through the masses of amyloid and at the periphery (8,9,25). In addition to clusters of plasma cells, Russell bodies and amyloid-containing foreign-body giant cells may be seen (63).
Pathogenesis. The amyloid is AL and is thought to be derived from local plasma cells. Isolation of plasma cell clones in the cutaneous infiltrates in some cases lends support to this theory, and some have suggested these lesions be considered a form of “low-grade B-cell lymphoproliferative disease” (60). Evidence that some of these lesions may be precipitated by antigenic induction in the setting of chronic inflammation is demonstrated by the close association to Sjögren syndrome in some cases (8,61).
Differential Diagnosis. On a histologic basis, differentiation of nodular amyloidosis from primary systemic amyloidosis is not possible.
Principles of Management. Techniques to improve the appearance of the lesion (corticosteroids, surgery, electrocautery, lasers, etc.) may be helpful, although lesions may recur.
COLLOID MILIUM AND NODULAR COLLOID DEGENERATION
Clinical Summary. There are four types of colloid degeneration of the skin: (a) juvenile colloid milium, (b) adult colloid milium, (c) nodular colloid degeneration, and (d) pigmented colloid milium. Juvenile colloid milium and nodular colloid degeneration are very rare, and pigmented colloid milium is exceedingly rare.
Juvenile colloid milium has its onset before puberty and shows numerous round or angular, brownish, waxy papules located mainly on sun-exposed areas, particularly the face (64,65). The lesions typically begin on the face and later develop on the posterior neck and dorsal hands (65). There is a family history in nearly half of the reported cases, and the possibility of a hereditary defect has been postulated (64,66). In some cases, ligneous conjunctivitis and ligneous periodontitis are associated findings (67).
Adult colloid milium, which starts in adult life, is clinically indistinguishable from the juvenile type, except in age of onset (Fig. 17-7). Sun exposure often seems to be a precipitating factor (65).
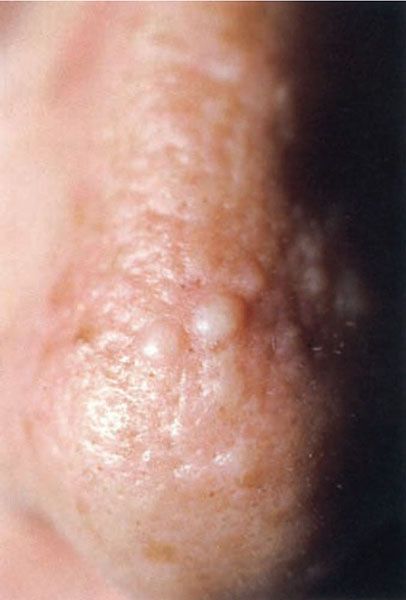
Figure 17-7 Colloid milium. These asymptomatic, whitish, soft papules on the nose were an incidental finding on examination. (From Elder DE, Elenitsas RE, Rubin AI, et al. Atlas and synopsis of Lever’s histopathology of the skin, 3rd ed. Philadelphia, PA: Lippincott Williams & Wilkins, 2013:302, with permission.)
Nodular colloid degeneration shows either a single large nodule on the face or multiple nodules on the face, chin, or scalp (68,69). Sun exposure does not seem to play a crucial role, because in some instances, the lesions are limited to the trunk, and a rare case involving the penis has been reported (69,70). Additionally, a case arising in herpes zoster scars has been reported (68).
Pigmented colloid milium is associated with exogenous ochronosis. This form occurs in areas subjected to application of hydroquinone, although exposure to chemical fertilizers was implicated in one case (71).
Histopathology. The colloid in the juvenile form is of epidermal origin and that in the other two forms is of dermal origin (64,69,72).
In juvenile colloid milium, fissured colloid masses fill the papillary dermis. The overlying epidermis is flattened with no grenz zone sparing, and basal cells show transformation into colloid bodies (65,67). Ultrastructurally, the colloid material is found within individual basal keratinocytes above an intact basement membrane and in extracellular aggregates in the dermis. These deposits contain remnants of nuclear membrane material, degenerating cell organelles, and desmosomes. The fibrillary structure is similar to that of amyloid ultrastructurally, although staining with Congo red sometimes fails to result in birefringence, and other stains for amyloid are often negative (64,67).
In adult colloid milium, a narrow zone of connective tissue usually separates the homogeneous masses of colloid located in the papillary dermis from the overlying epidermis (Fig. 17-8). Elastic tissue staining shows some elastic fibers in this narrow zone of connective tissue. In addition, solar elastotic fibers are usually seen in the background and at the base of the colloid deposits. Cleft-like spaces containing fibroblasts may be present within the deposits. The colloid deposits in adult colloid milium are usually weakly positive with Congo red and may display weak green birefringence (72).
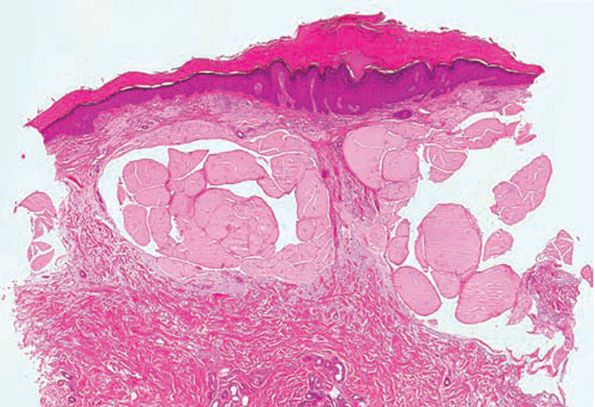
Figure 17-8 Colloid milium, adult type. Homogeneous, fissured masses of colloid are present in the uppermost dermis. The colloid material greatly resembles the amyloid material observed in primary systemic amyloidosis (see Fig. 17-2, page 503).
In nodular colloid degeneration, the epidermis is flattened. The upper three quarters of the dermis are filled with pale pink homogeneous material; in some lesions, even the entire dermis is filled with this material (73). A grenz zone is sometimes present. Scattered nuclei of fibroblasts are present within the colloid. There are scattered clefts or fissures and some dilated capillaries. The hair follicles and sebaceous glands appear well preserved (69).
Pigmented colloid milium is similar to adult colloid milium but contains areas of yellow-brown pigmentated globules similar to ochronosis (71).
Pathogenesis. Colloid shows considerable resemblance to amyloid—not only in its histologic appearance but also in its histochemical reactions (72).
Colloid, like amyloid, is periodic acid–Schiff (PAS) positive and diastase resistant. Staining with Congo red results in green birefringence (sometimes weak), and the colloid fluoresces after staining with thioflavin T. However, it does not react with pagoda red and other cotton dyes (9,72). Serum amyloid P not only is a component of normal and abnormal dermal elastic fibers but reportedly can be stained immunocytochemically in adult colloid milium and nodular colloid degeneration (74,75). Juvenile colloid milium, however, exhibits a positive immunostaining reaction to polyclonal antikeratin antibody. This suggests that the histogenesis of this process may differ from that of the adult form, in that the colloid in juvenile colloid milium is more likely derived from keratinocytes and related to amyloid K (75).
Electron microscopy in a case of juvenile colloid milium has shown that the colloid consists of tightly packed bundles of filaments, 8 to 10 nm thick, in a wavy or whorled arrangement (67,75,76). The colloid, which forms by filamentous degeneration of tonofilaments, maintains a cytoid configuration in the dermis and may contain nuclear remnants (67,76).
In adult colloid milium, the colloid masses are seen on electron microscopic examination to consist primarily of a granulofibrillar, amorphous substance. On high magnification, very fine, branching, wavy filaments, only 2 nm wide, may be seen embedded in the amorphous substance (72,74). There is rather conclusive evidence that colloid is derived from elastic fibers through sequential degenerative changes. This can be observed at the periphery of the lesion, where colloid is being produced, and within the colloid, where fibrils with a tubular structure and a diameter of 10 nm that are strikingly similar to the microfibrils of elastic fibers can be seen (77). The colloid in adult colloid milium thus represents a final product of severe solar degeneration (72,75).
The colloid in nodular colloid degeneration, similar to that in adult colloid milium, consists of an amorphous substance and short, wavy, irregularly arranged filaments (73). The diameter of these filaments is 3 to 4 nm (68). These electron microscopic findings exclude nodular amyloidosis as the diagnosis, because in this condition, the filaments have a diameter of about 6 to 7 nm and are long and straight.
Principles of Management. Dermabrasion, cryotherapy, and other modalities have been used with limited success.
LIPOID PROTEINOSIS (HYALINOSIS CUTIS ET MUCOSAE/URBACH–WIETHE DISEASE)
Clinical Summary. Lipoid proteinosis is a rare autosomal recessive disorder first described by Siebenmann in 1908. It was established as a distinct clinical and histologic entity by Urbach and Wiethe in 1929. In 1932, Urbach coined the term lipoid proteinosis (78).
Clinically, there are papular and nodular lesions on the face. Areas of diffuse infiltration associated with hyperkeratosis are observed on the elbows, knees, hands, and occasionally elsewhere. The papules and nodules on the face cause pitted, pock-like scars, giving the skin a pigskin-leather appearance. Verrucous plaques can form in areas of friction (79). Beads of small nodules may be present along the free margins of the eyelids. Alopecia may be present from scalp involvement (79,80). The tongue is firm because of diffuse infiltration. Two of the hallmark findings are extension of the infiltration to the frenulum of the tongue, restricting its movement, and infiltration of the vocal cords, causing hoarseness (81). Seizures, an abnormal perception and appraisal of fear, and other neuropsychiatric symptoms are sometimes manifest (80,81).
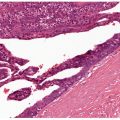
Histopathology. There is extensive cutaneous deposition of amorphous eosinophilic material surrounding capillaries and sweat coils and in the thickened papillary dermis (Fig. 17-9). Focal deposits are found in the deeper dermis. In verrucous lesions, the homogeneous bundles often are oriented perpendicular to the skin surface. This hyaline material is PAS positive and resistant to diastase digestion. Staining with Alcian blue at pH 2.5 is slightly positive and is sensitive to hyaluronidase digestion. The presence of lipids, highlighted with oil red O and sudan black stains, is variable and likely results from adherence of lipids to glycoproteins rather than from abnormal lipid production (82).
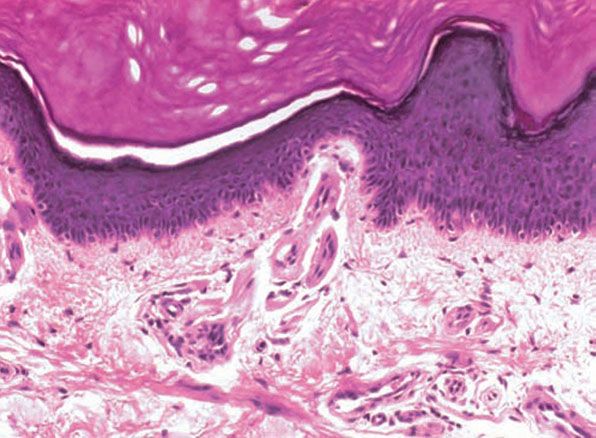
Figure 17-9 Lipoid proteinosis. The hyaline material consists of thick mantles surrounding the blood vessels.
Systemic Lesions. Intracranial calcification noted frequently in brain imaging of patients with lipoid proteinosis has been held responsible for seizure activity (79,80,82). Autopsy findings have established that the calcium is deposited within the walls of capillaries located in the hippocampal gyri of the temporal lobes (83). The fact that after decalcification a mantle of PAS-positive material is observed around the endothelium of these vessels indicates that hyalinization precedes calcification of the capillary walls (84).
The widespread distribution of the pericapillary hyalin deposits has been established by biopsies and autopsies. Deposits have been found in the submucosae of the gastrointestinal and upper respiratory tracts and in the vagina; in the retina between the vitreous membrane and the pigment epithelium; and in the testes, pancreas, lungs, kidneys, and elsewhere (80,82,84).
Pathogenesis. Two different substances present in lesions of lipoid proteinosis have the light microscopic appearance of hyalin because they consist of homogeneous eosinophilic material and are PAS positive and diastase resistant. However, their different appearances and origins become evident on electron microscopy (82,85). One represents hyalin-like material consisting of multiplications of basal laminae, produced by a variety of cells. The other is true hyalin, and different studies have suggested this material is produced by fibroblasts, as denoted by distinct cytoplasmic vacuoles, or by dermal eccrine glands and histiocytes (79,80,86). Abnormal lysosomes with curved tubular profiles in eccrine glands and histiocytes may reflect a defect in the degradation pathway of glycolipids and sphingolipids, similar to lysosomal storage diseases such as Faber disease (79,80). Others have suggested that defective cutaneous lymphatics or microvascular abnormalities may play a role (86).
Lipids are not an essential feature of the disease, and there is variability in the type of lipid and amount present. They can be removed with lipid solvents without damage to the protein–carbohydrate complex of the true hyalin, suggesting they are either free or loosely bound to the hyalin (82,85).
Mutations in the extracellular matrix protein 1 (ECM1) gene have been identified as the cause of lipoid proteinosis (87). The glycoprotein product of this gene is thought to play a functional role in cutaneous physiology and homeostasis. With loss of function of ECM1, there is an accumulation of lipid-like materials in various viscera as well as in the skin, as previously described (82,85). ECM1 appears to have many functions, although loss of its function in binding of key components of dermal ground substance and regulation of the bioactivity of matrix components, such as matrix metalloproteinase 9, appears to have a crucial role in the development of lipoid proteinosis (79).
On electron microscopic examination, three major alterations are noted in the dermis: (a) a considerable thickening of basal laminae; (b) massive depositions of amorphous material (hyalin), predominantly in the upper dermis and around blood vessels; and (c) a marked reduction in the number and size of collagen fibrils (79,80). The thickening of the basal laminae in multiple layers is evident around small blood vessels, skin appendages, smooth muscle cells, perineuria, and Schwann cells. In contrast, there is almost no multiplication of the basal lamina of the epidermis (88). Around capillaries, basal laminae are several layers thick, arranged in a concentric “onion-skin” arrangement (80,82). Interspersed between these layers are fine collagen fibrils in an amorphous matrix. The thickened basement membranes stain intensively for laminin and collagen types IV and VII (80,85).
A unique feature of hyalinosis cutis et mucosae consists of large depositions of amorphous material in the dermis. The exact chemical nature of this hyaline material is not known, but it is a noncollagenous glycoprotein that contains neither laminin nor fibronectin (89).
The collagen within the areas of hyalin deposits appears as fine but otherwise normal fibrils arranged in bundles or in random fashion. Most fibrils have a diameter less than 50 nm, compared with the usual size of 70 to 140 nm. Involved skin shows a fivefold reduction in collagen type I, the major collagen species of normal skin, and to a lesser extent in collagen type III, as indicated by cell culture studies. As judged by the contents of glycine and hydroxyproline, normal skin contains about 80% collagen, but involved skin in lipoid proteinosis contains only 20% collagen as the result of a large accumulation of noncollagenous glycoproteins.
Cultured fibroblasts from lesions of lipoid proteinosis have shown an increased synthesis of noncollagenous proteins at the expense of newly synthesized collagens. The hyaline material in lipoid proteinosis originates from the overproduction of noncollagenous proteins, most of which are normal constituents of human skin (80,89).
Differential Diagnosis. Porphyria shows deposits of hyalin-like material around the superficial dermal capillaries that are usually less dense but otherwise indistinguishable from the perivascular deposits of hyalin-like material seen in lipoid proteinosis. However, involvement of the membrana propria of the sweat glands is rare, and no true dermal hyalin is present. In addition, the cutaneous lesions in porphyria are limited to sun-exposed areas.
Principles of Management. Management depends on organ systems involved, and may include dermabrasion for cutaneous lesions and surgical removal of vocal cord polyps.
PORPHYRIA
Clinical Summary. Eight types of porphyria are recognized, each caused by abnormalities in the porphyrin synthesis pathway and with varied clinical features (Table 17-1). The light sensitivity in the six types with cutaneous lesions is caused by wavelengths that are absorbed by the porphyrin molecule. These wavelengths lie in the 400-nm range, representing long-wave ultraviolet light (UVA) and visible light (90).
In erythropoietic porphyria (Günther disease), a very rare disease that typically develops during infancy or childhood, recurrent vesiculobullous eruptions in sun-exposed areas of the skin gradually result in ulcerations and scarring. Repeated scarring and infections in these areas can lead to disfigurement. Areas of hypo- and hyperpigmentation may also be seen. Hydrops fetalis may occur in affected embryos. Hypertrichosis, red fluorescent urine, and reddish-brown stained teeth that fluoresce are additional features (90,91).
In erythropoietic protoporphyria, the usual reaction to light is pruritic and burning erythema and edema followed by thickening and superficial scarring of the skin. The skin may take on a waxy or leathery appearance, particularly on the face and hands (92,93). In rare instances, vesicles are present that may resemble those seen in hydroa vacciniforme (94,95). The protoporphyrin is formed in reticulocytes in the bone marrow and is then carried in circulating erythrocytes and in the plasma. When a smear of blood from a patient is examined under a fluorescence microscope, large numbers of red-fluorescing erythrocytes are observed. The protoporphyrin is cleared from the plasma by the liver and excreted into the bile and feces (91). It is not found in the urine (95). In 1% to 4% of patients, fatal liver disease develops quite suddenly, usually in persons of middle age (96) but occasionally in patients only in the second decade of life (97,98).
In porphyria variegata, different members of the same family may have either cutaneous manifestations identical to those of porphyria cutanea tarda (PCT) or systemic involvement analogous to acute intermittent porphyria, or both, or the condition may remain latent. It is caused by a deficiency in the activity of protoporphyrinogen oxidase (99–101). The presence of protoporphyrin in the feces distinguishes porphyria variegata from PCT. Also, a sharp fluorescence emission peak at 626 nm is specific for the plasma of porphyria variegata (102).
Three forms of PCT can be distinguished: sporadic, familial, and hepatoerythropoietic (103). In the sporadic form (type I), only the hepatic activity of uroporphyrinogen decarboxylase is decreased. Almost all patients are adults, and no clinical evidence of PCT is found in other members of the patient’s family. Although the sporadic form can occur without any precipitating factor, in most instances, in addition to the inherited enzymatic defect, an acquired damaging factor to liver function is needed. Most commonly this is ethanol, but iron supplements, estrogen treatment, and hepatitis are also frequently implicated (104).
The familial form is subdivided into types II and III, and is a dominantly inherited disorder with low penetrance. In type II, in addition to the hepatic activity, the extrahepatic activity of uroporphyrinogen decarboxylase is decreased to about 50% of normal. The enzymatic activity usually is determined on the erythrocytes. Type III is very rare, and the decreased uroporphyrinogen decarboxylase activity is limited to hepatocytes. The familial form may occur at any age, including childhood, and often, but not always, there is a family history of overt PCT (104). A reliable laboratory method for the diagnosis of the porphyrias is now provided by molecular genetics (105,106).
In the very rare hepatoerythropoietic form, the skin lesions appear in childhood and the activity of uroporphyrinogen decarboxylase in all organs is decreased to 3% to 27% of normal. Family studies suggest that the inheritance pattern in these patients is autosomal recessive and they are either homozygous for the gene that causes PCT or compound heterozygous (103,107).
Clinically, the sporadic form of PCT, by far the most common type of porphyria, shows blisters that arise through a combination of sun exposure and minor trauma, mainly on the dorsa of the hands but sometimes also on the face (Fig. 17-10). Mild scarring commonly results with occasional milia. The skin of the face and the dorsa of the hands are often thickened and sclerotic. Hypertrichosis of the face is common. Evidence of hepatic cirrhosis with siderosis is regularly present but generally is mild (105). In rare instances, hepatocellular carcinoma or a carcinoma metastatic to the liver induces PCT (108,109). In the familial form of PCT, the clinical picture is similar to that of the sporadic form, but the changes are more pronounced. In the hepatoerythropoietic form, the manifestations are even more severe. On clinical grounds, the symptoms of most patients resemble those of erythropoietic porphyria, but when symptoms are milder, they resemble those of erythropoietic protoporphyria (110). Vesicular eruptions lead to ulceration, severe scarring, partial alopecia, and sclerosis (107,111,112). Erythrocytes and teeth may show fluorescence (107). Liver damage develops with increasing age (90,113,114). Arthritis is a rare complication (107).
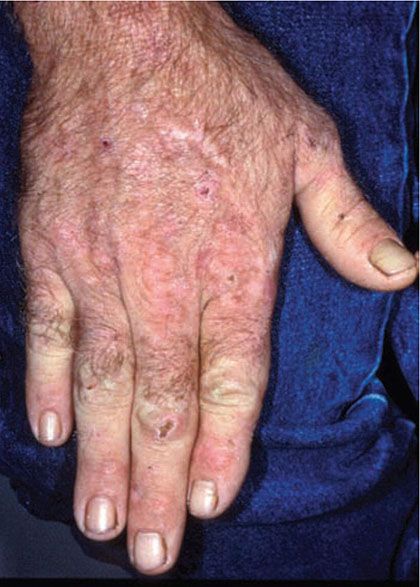
Figure 17-10 Porphyria cutanea tarda. There are crusts from healing vesicles scattered over the dorsal hand. Scars and milia are present on the mid-dorsal hand.
In hereditary coproporphyria, a very rare disorder, there are episodic attacks of abdominal pain and a variety of neurologic and psychiatric disturbances analogous to those observed in acute intermittent porphyria and porphyria variegata (90,115). In some cases, there are also cutaneous manifestations indistinguishable from those of PCT and porphyria variegata (90,115).
Histopathology. The histologic changes in the skin lesions are the same in all six types of porphyria with cutaneous lesions. Differences are based on the severity rather than on the type of porphyria. Homogeneous, eosinophilic material is regularly observed, and bullae are present in some instances (90,116,117). In addition, sclerosis of the collagen is present in old lesions (117,118).
In mild cases, homogeneous, pale, eosinophilic deposits are limited to the immediate vicinity of the blood vessels in the papillary dermis (90,116,117). These deposits are best visualized with a PAS stain, being PAS positive and diastase resistant (90,117).
In severely involved areas, which are most common in erythropoietic protoporphyria, the perivascular mantles of homogeneous material are wide enough in the papillary dermis to coalesce with those of adjoining capillaries. In addition, deeper blood vessels may show homogeneous material around them, and similar homogeneous material may be found occasionally around eccrine glands (119). PAS staining demonstrates this material particularly well. In some instances, it also contains acid mucopolysaccharides, shown with Alcian blue or the colloidal iron stain (120), or lipids, demonstrable with Sudan IV or Sudan black B (119,121). In addition, the PAS-positive dermal–epidermal basement membrane zone may be thickened (117,120).
In areas of sclerosis, which occur especially in PCT, the collagen bundles are thickened. In contrast to scleroderma, PAS-positive, diastase-resistant material is often present in the dermis in perivascular locations (120).
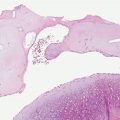
The bullae, which are most common in PCT and least common in erythropoietic protoporphyria, arise subepidermally (Fig. 17-11). Some blisters are dermolytic and arise beneath the PAS-positive basement membrane zone, below the sublamina densa (117); others form in the lamina lucida and are situated above the PAS-positive basement membrane zone (117,122) (Fig. 17-12). Some demonstrate bullous pemphigoid antigen, laminin, and types IV and VII collagen on both sides of the blister, and thus do not show a defined level of cleavage, whereas others show collagen IV on the roof and collagen VII on both sides, and therefore the level of cleavage is at the level of the sublamina densa. Other cases show all antigens below the plane of cleavage and are intraepidermal (117). Thus, there is no characteristic level of cleavage. It has been suggested that the blisters in PCT in mild cases arise within the junctional zone but that in severe cases they form beneath the PAS-positive basement membrane zone and thus heal with scarring (123). It is quite characteristic of the bullae of PCT that the dermal papillae often extend irregularly from the floor of the bulla into the bulla cavity (117). This phenomenon, referred to as festooning, is explained by the rigidity of the upper dermis induced by the presence of eosinophilic material within and around the capillary walls in the papillae and the papillary dermis.

Figure 17-11 Porphyria cutanea tarda. A: There is a subepidermal blister. The architecture of the dermal papillae remains intact. The dermis contains no significant inflammatory infiltrate (H&E). B: Vessels within preserved dermal papillae at the blister base are surrounded by PAS-positive, diastase-resistant deposits.

Figure 17-12 Porphyria cutanea tarda. On staining with the PAS reaction after diastase digestion, the PAS-positive basement membrane zone is observed at the floor of the blister. PAS-positive hyalin-like material is present in the walls of capillaries in the upper dermis.
The epidermis forming the roof of the blister often contains eosinophilic bodies that are elongate and sometimes segmented (124). These “caterpillar bodies” are PAS positive and diastase resistant. Ultrastructurally, they have been found to contain three components: (a) cellular organelles, including melanosomes, desmosomes, and mitochondria; (b) colloid that may be located intracellularly or extracellularly; and (c) electron-dense material thought to be of basement membrane origin (125). While fairly specific, these structures are not always present, and other conditions may demonstrate “caterpillar body–like” clusters (124).
Pathogenesis. The substance around dermal vessels has the appearance of hyalin because it consists of homogeneous, eosinophilic material and is PAS positive and diastase resistant. However, just as the perivascular material in lipoid proteinosis, it is only hyalin-like and consists of multiplications of basal laminae (116). True hyalin is absent.
On electron microscopic examination, concentric duplications of the basement membrane around the dermal blood vessels are observed. Peripheral to this multilayered basement membrane, one observes a thick mantle of unlayered material with the same filamentous and amorphous composition as that of the basement membrane (117). Often, a gradual transition from the layered to the unlayered zone can be observed (126,127). Scattered through the thick, unlayered zone are solitary collagen fibrils with an average diameter of only 35 nm, in contrast to the 100 nm of mature collagen (126,128). In cases with severe involvement, presumably from repeated injury to the vessels, intermingled filamentous and amorphous material is seen throughout the upper dermis and even in the mid-dermis.
As the perivascular material in porphyria represents excessively synthesized basement membrane material, it demonstrates immunofluorescence staining with anti–type IV collagen antibody (117). The presence of small collagen fibrils analogous to reticulum fibrils suggests the presence also of type III collagen (129). Tryptophan, a substance derived from blood and not present in collagen or elastic tissue, is present in the hyalin-like deposits, suggesting that the material is also derived from the vessel wall and leakiness of its contents (117).
Direct immunofluorescence studies have demonstrated deposition of immunoglobulins, particularly immunoglobulin G (IgG) and complement C3, and to a lesser extent IgA and IgM, in the vessel walls and at the dermal–epidermal junction of light-exposed skin. IgG is more persistent throughout inactive and treated phases of the disease (117). It has been deemed unlikely by some that these deposits indicate an immunologic phenomenon and that rather, they are the result of “trapping” of immunoglobulins and complement in the filamentous material. Observations of conditions where the complement cascade has been inactivated or inhibited, however, suggest that complement activation may play a key role in the development of endothelial injury in cutaneous lesions of porphyria (117,130).
The enzymatic defect in each form of porphyria is known (see Table 17-1). Enzyme determinations may be carried out on cultured skin fibroblasts, erythrocytes, or liver tissue.
Liver damage is generally mild and chronic in PCT. In erythropoietic protoporphyria, however, liver function tests are usually normal, even though microscopic deposits of protoporphyrin in the liver are found frequently (92,95). Yet, in rare instances, death occurs from liver failure developing swiftly after the initial detection of hepatic dysfunction (131). Patients with liver failure have very high levels of protoporphyrin in the erythrocytes and at the time of death show extensive deposits of protoporphyrin in a cirrhotic liver. The protoporphyrin in hepatocytes and Kupffer cells appears as dark brown granules (131). This pigment exhibits birefringence on polariscopic examination and in unstained sections viewed with ultraviolet light shows red autofluorescence (131). In patients with normal liver function tests, biopsy of the liver may or may not show portal and periportal fibrosis (132).
Principles of Management. Discontinuance of risk factors (alcohol, offending drugs, iron supplements, etc.) is recommended. Administration of hemin aids in acute attacks. Avoidance of sunlight and phlebotomy are mainstays of treatment for cutaneous lesions. Antimalarial therapy may be used as an adjunctive measure in some cases. Abdominal, cardiac, and nervous system manifestations are treated symptomatically. Acute intermittent porphyria may be treated with hemin.
Pseudoporphyria Cutanea Tarda
Clinical Summary. In patients with chronic renal failure who are receiving maintenance hemodialysis, an eruption indistinguishable from that of PCT may develop on the dorsa of the hands and fingers during the summer months (133). Rarely, blisters are seen also on the face, and atrophic scarring develops (134). Large case series of patients receiving hemodialysis have shown that 1% to 18% of patients showed this type of eruption (135–137). Normal porphyrin levels in urine, stool, and plasma are the rule in hemodialyzed patients developing clinical signs of PCT, although a true PCT may coexist (137,138). In such patients, if a certain degree of diuresis persists, urinalysis may not be representative of the porphyria metabolism, and the plasma and fecal porphyrins should always be measured (139).
PseudoPCT may also occur following the ingestion of certain drugs, such as naproxen, furosemide, nalidixic acid, tetracycline hydrochloride, hydrochlorothiazide, retinoids, voriconazole, cyclosporine, and oral contraceptives (140–143). Because many patients on hemodialysis for renal failure also are receiving furosemide, withdrawal of the drug can determine whether the dialysis or the medication is the cause of the pseudoporphyria, because in drug-induced cases, withdrawal of the drug is curative (144,145).
Histopathology. In patients with pseudoporphyria, the histologic picture is indistinguishable from that seen in mild cases of porphyria. The superficial blood vessels show thickened walls, and the PAS-positive basement membrane zone is often thickened as well. Blisters are subepidermal, with festooned dermal papillae. The blisters usually are situated above the PAS-positive basement membrane zone (134,135,139,141,144).
Pathogenesis. Electron microscopic findings are identical to those of porphyria (135). As in porphyria, immunoglobulins are often observed in vessel walls and at the dermal–epidermal junction. Complement is also occasionally present (133–135).
Principles of Management. For hemodialysis-associated cases, patients may be treated with N-acetylcysteine. Discontinuation of the offending agents suffices in drug-induced cases. Sun protection is recommended.
CALCINOSIS CUTIS
There are four forms of calcinosis cutis: metastatic calcinosis cutis, dystrophic calcinosis cutis, idiopathic calcinosis cutis, and subepidermal calcified nodule.
Metastatic Calcinosis Cutis
Clinical Summary. Metastatic calcification develops as the result of hypercalcemia or hyperphosphatemia. Hypercalcemia may result from (a) primary hyperparathyroidism, (b) excessive intake of vitamin D, (c) excessive intake of milk and alkali (146), or (d) extensive destruction of bone through by metastases of a carcinoma (147). Other associated conditions reported include alcoholic liver disease, systemic lupus erythematosus, rheumatoid arthritis, diabetes, protein C or S deficiency, and Crohn disease (147–149). Hyperphosphatemia occurs in chronic renal failure as the result of a decrease in renal clearance of phosphorus and is associated with a compensatory drop in the serum calcium level. The low level of ionized calcium in the serum stimulates parathyroid secretion, leading to secondary hyperparathyroidism and to resorption of calcium and phosphorus from bone. The demineralization of bone causes both osteodystrophy and metastatic calcification (150).
Metastatic calcification most commonly affects the media of the arteries, periarticular regions, and the kidneys. In addition, other visceral organs, such as the myocardium, the stomach, and the lungs may be involved (151). Within the vessels, this is primarily thought to occur by osteoblastic transdifferentiation of vascular smooth muscle cells in response to uremic serum, hyperphosphatemia, and hypercalcemia (150,152).
Metastatic calcification in the subcutaneous tissue is occasionally observed in association with renal hyperparathyroidism (153), in uremia (154), in hypervitaminosis D (146), and as the result of excessive intake of milk and alkali (146), but rarely in primary hyperparathyroidism (155,156). Palpable, hard nodules, occasionally of considerable size, are located mainly in the vicinity of the large joints (146,157). With increasing size, the nodules may become fluctuant (158).
Calciphylaxis, or calcific uremic arteriolopathy, is a life-threatening condition in which there is progressive calcification of small- and medium-sized vessels of the subcutis often accompanied by necrosis (see also Chapter 8). It most frequently arises in the setting of hyperparathyroidism associated with chronic renal failure. Calciphylaxis is often associated with an elevated serum calcium/phosphate product, with other factors, including several glycoproteins (matrix G1a protein and glycopontin) likely playing a role in the development of vascular calcification (159).
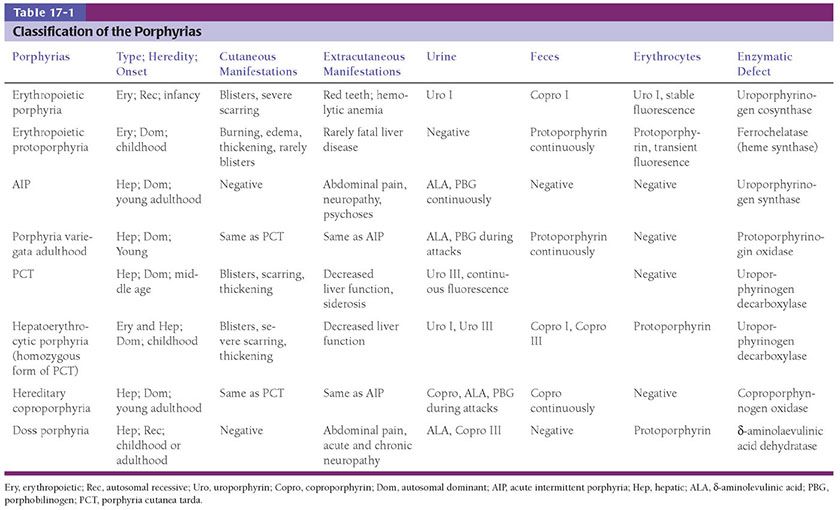
Clinically, the lesions present as a panniculitis or vasculitis. Bullae, ulcerations, or a livedo reticularis–like eruption can be present (Fig. 17-13) (146,149). Potential complications of gangrene, sepsis, pancreatitis, and multisystem organ failure contribute to an overall mortality of 60% to 80% (160,161).
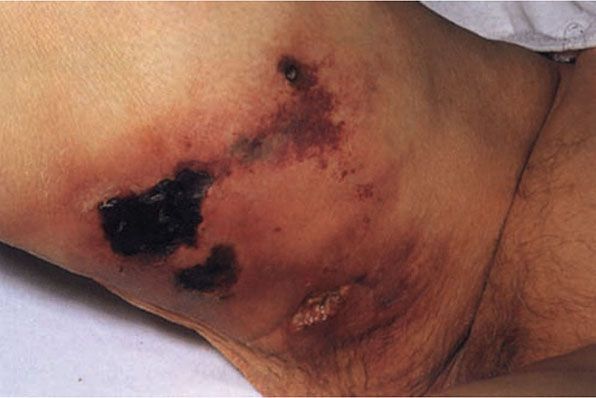
Figure 17-13 Calciphylaxis. This elderly female with end-stage renal disease and elevated parathyroid hormone level developed widespread induration of the lower extremities which led to purpuric and necrotic ulcerations. (From Elder DE, Elenitsas RE, Rubin AI, et al. Atlas and synopsis of Lever’s histopathology of the skin, 3rd ed. Philadelphia, PA: Lippincott Williams & Wilkins, 2013:232, with permission.)
Instances of cutaneous metastatic calcinosis are rare. Most reports have concerned patients with renal hyperparathyroidism and osteodystrophy. The cutaneous lesions may consist of tender, firm, white to erythematous papules, nodules, or plaques (162,163). Papules in a linear arrangement (158) and symmetric, nodular plaques may occur (164). The lesions may ulcerate and a chalky, white substance can be expressed (162).
Mural calcification of arteries and arterioles in the deep dermis or subcutaneous tissue occurs rarely in primary hyperparathyroidism (147), but somewhat more frequently in secondary hyperparathyroidism subsequent to renal disease, particularly if subsequent to dialysis for chronic renal disease or to renal allograft (147,165). This may lead to occlusion of these vessels and to infarctive ulcerations, especially on the legs.
Histopathology. Calcium deposits are recognized easily in histologic sections, because they stain deep blue with H&E (hematoxylin and eosin). They stain black with the von Kossa stain for calcium phosphate. As a rule, the calcium occurs as massive deposits when located in the subcutaneous fat and usually as granules and small deposits when located in the dermis (Fig. 17-14). Large deposits of calcium often evoke a foreign-body reaction; thus, giant cells, an inflammatory infiltrate, and fibrosis may be present around them (166,167).
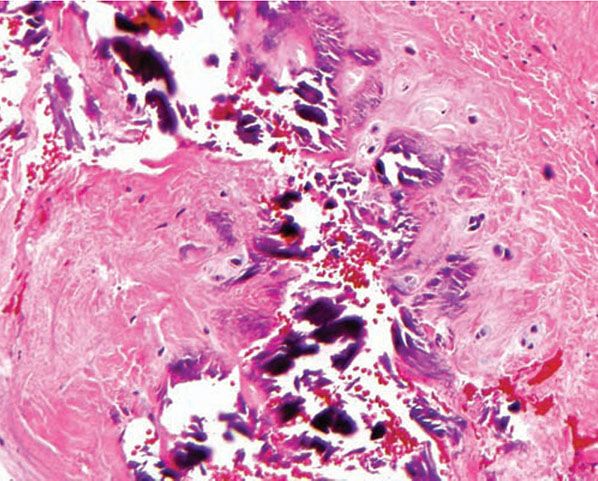
Figure 17-14 Metastatic calcinosis cutis. Amorphous deposits of basophilic material are present in the dermis.
In areas of infarctive necrosis, as a result of calcification of dermal or subcutaneous arteries or arterioles, the involved vessels show calcification of their walls and intravascular fibrosis with attempts at recanalization of the obstructed lumina (159,167). Mural calcification often is most pronounced in the internal elastic membranes of arteries or arterioles (167).
The histologic changes in calciphylaxis include calcium deposits in the subcutis, chiefly within the walls of small- and medium-sized vessels (Fig. 17-15). These deposits can be associated with endovascular fibrosis, thrombosis, or global calcific obliteration (Fig. 17-16) (167). The fully evolved disease process shows, in addition, areas of necrosis with a clean background or accompanied by neutrophils (166,168).
Figure 17-15 Calciphylaxis. There is a panniculitis with associated calcification of soft tissues in the walls of small vessels. (A: H&E, original magnification ×20; B: H&E, original magnification ×100.)

Figure 17-16 Calciphylaxis, high power. The calcification affects the media of this small artery. In the same vessel, the intima is thickened by delicate fibroplasia, greatly compromising the lumen. The intimal fibrosis has retracted from the stiffened calcified wall due to fixation artifact. (From Elder DE, Elenitsas RE, Rubin AI, et al. Atlas and synopsis of Lever’s histopathology of the skin, 3rd ed. Philadelphia, PA: Lippincott Williams & Wilkins, 2013:232, with permission.)
It is particularly important that these findings be recognized in order that appropriate therapy might be instituted immediately.
Principles of Management. Treatment is aimed at reducing serum calcium and phosphate levels and may include calcimimetics, bisphosphanates, and parathyroidectomy. Intravenous immunoglobulin (IVIg) has been reported to be beneficial in rare case reports. Use of high-frequency ultrasound and sodium thiosulfate is investigational at this time.
Dystrophic Calcinosis Cutis
Clinical Summary. In dystrophic calcinosis cutis, the calcium is deposited in previously damaged tissue. The values for serum calcium and phosphorus are normal, and the internal organs are spared. There may be numerous large deposits of calcium (calcinosis universalis) or only a few deposits (calcinosis circumscripta).
Calcinosis universalis occurs as a rule in patients with dermatomyositis (Fig. 17-17), but exceptionally it has also been observed in patients with systemic scleroderma, systemic lupus erythematosus, and connective tissue overlap diseases, and it may follow infections or trauma (169,170). Exceedingly rare cases may be preceded by the onset of an underlying connective tissue disorder (171). Large deposits of calcium are found in the skin and subcutaneous tissue and often in muscles and tendons (172). In dermatomyositis, if the patient survives, the nodules of dystrophic calcinosis gradually resolve.

Figure 17-17 Dystrophic calcinosis cutis. Granules and globules of calcium are located beneath the epidermis. This biopsy is from the buttock of a child with dermatomyositis who had extensive calcification also of muscles and tendons.
Calcinosis circumscripta occurs as a rule in patients with systemic scleroderma; rarely, however, it may be observed in patients with widespread morphea (173,174). Generally, in the presence of calcinosis, systemic scleroderma manifests itself as acrosclerosis. The association of acrosclerosis and calcinosis is often referred to as the Thibierge–Weissenbach syndrome or as the CREST syndrome, because the manifestations usually consist of calcinosis cutis, Raynaud phenomenon, esophageal dysfunction, sclerodactyly, and telangiectasia (146,175). Patients with this syndrome often have a better prognosis than those with generalized scleroderma or systemic sclerosis. Clinically, calcinosis circumscripta shows successively appearing areas of induration that often break down and extrude white, chalky material.
In addition to occurring in the connective tissue diseases, dystrophic calcinosis is often seen in subcutaneous fat necrosis of the newborn and, rarely, in inherited disorders such as Ehlers–Danlos disease, Werner syndrome, and pseudoxanthoma elasticum (176).
Histopathology. As in metastatic calcinosis cutis, the calcium in dystrophic calcinosis cutis usually is present as granules or small deposits in the dermis and as massive deposits in the subcutaneous tissue. A foreign-body giant cell reaction with inflammation and fibrosis is often found around large deposits of calcium (146). The calcium deposits usually are located in areas in which the collagen or fatty tissue appears degenerated as a result of the disease preceding the calcinosis.
Principles of Management. Treatment is guided at pain relief and cosmesis. Intralesional corticosteroids have limited benefit, and etidronate disodium, aluminum hydroxide, low-dose anticoagulants, bisphosphonates have shown slightly better results. Resolution of lesions may be seen with topical sodium thiosulfate. Surgical removal may be required for severe cases and when lesions impair function, although recurrence or new lesions may result.
Idiopathic Calcinosis Cutis
Clinical Summary. Even though the underlying connective tissue disease in some instances of dystrophic calcinosis cutis may be mild and can be overlooked unless specifically searched for, there remain cases of idiopathic calcinosis cutis that resemble dystrophic calcinosis cutis but show no underlying disease (173,177,178).
One entity is regarded as a special manifestation of idiopathic calcinosis cutis: tumoral calcinosis. It consists of solitary or numerous large, subcutaneous, calcified masses, generally along extensor surfaces of large joints, which may be associated with papular and nodular skin lesions of calcinosis (176,179,180). Dental manifestations from calcifications of the pulp may be present (179). The disease usually is familial and is associated with hyperphosphatemia (176,179). Recent studies have elucidated the genetic basis of familial tumoral calcinosis to be caused by mutations in three genes involved in fibroblast growth factor 23 function: FGF23, KL, and GALNT3 (179,181). Otherwise, the similarities of tumoral calcinosis to the dystrophic calcinosis universalis observed with dermatomyositis are marked.
Histopathology. Tumoral calcinosis shows subcutaneous or intramuscular large masses of calcium surrounded by a foreign-body reaction (146,176,179,182). Intradermal aggregates are present in some cases (146,179). Discharge of calcium may take place through areas of ulceration or by means of transepidermal elimination (146,180). The inactive phase of the disease typically shows dense fibrous septa without cellular elements (179,183).
Pathogenesis. Lesions of idiopathic calcinosis cutis studied by electron microscopy show that the deposits consist of pleomorphic calcium phosphate (hydroxyapatite) crystals (182). These deposits of calcium have been observed within collagen fibrils and situated in the ground substance (184,185). Many ultrastructural studies have implicated intramitochondrial calcification in histiocytes as the principal calcifying process in tumoral calcinosis (182,186).
Principles of Management. Surgical excision combined with phosphate deprivation and acetazolamide has proven to be effective.
Idiopathic Calcinosis of the Scrotum
Clinical Summary. Idiopathic calcinosis of the scrotum consists of multiple asymptomatic nodules of the scrotal skin. The nodules usually begin to appear in the third decade of life, although onset as early as 9 years old or as late as 85 years old has been reported. They increase in size and number, are rarely pruritic, and sometimes break down to discharge their chalky contents (187).
Histopathology. Originally, the accepted view was that some of the calcific masses in calcinosis of the scrotum were surrounded by a granulomatous foreign-body reaction and others were not (188). However, in recent studies in which numerous scrotal nodules were examined at different times in the same patients, some of the smaller lesions were epidermal cysts, whereas other cystic lesions showed calcification of their keratin contents, and still others showed ruptures of their epithelial walls. The cyst wall seems to eventually be destroyed, leaving only dermal collections of calcium that can become larger. Thus, according to this view, calcinosis of the scrotum represents the end stage of dystrophic calcification of scrotal epidermal cysts (188). It can be assumed that many early lesions start out as cysts but, as they age and calcify, lose their cyst walls. In some cases, eccrine duct milia have been favored as the origin because of positive reactions for carcinoembryonic antigen and epithelial membrane antigen, markers of eccrine differentiation (189). Some reports have alternatively suggested dartoic muscle degeneration as a possible etiology (190). It is likely that these lesions are commonly derived from epidermal cysts, although the cause may potentially be multifactorial, and in cases without definitive pathogenesis, designation as idiopathic remains appropriate.
Principles of Management. Treatment is limited to surgical excision.
Subepidermal Calcified Nodule
Clinical Summary. In subepidermal calcified nodule, also referred to as cutaneous calculi, usually a single, small, raised, hard nodule is present, usually on the face. Occasionally, however, there are two or three nodules, and in some instances, there are numerous or even innumerable nodules. Most patients are children; however, in some patients, a nodule is present at birth or does not appear until adulthood (191). A male predominance with a ratio of 2:1 has been observed (192). In most instances, the surface of the nodule is verrucous, but it may be smooth, and lesions may mimic verruca vulgaris or molluscum contagiosum (192,193).
Histopathology. The calcified material is located predominantly in the uppermost dermis, although in large nodules, it may extend into the deep layers of the dermis. The calcium is present largely as closely aggregated globules (Fig. 17-18). In some instances, however, there are also one or several large, homogeneous masses of calcified material (192,194). Both the globules and the homogeneous masses occasionally contain well-preserved nuclei (194). A lymphohistiocytic infiltrate, macrophages, and foreign-body giant cells may be arranged around the large, homogeneous masses (192,194). The epidermis is often hypertrophic. Calcium granules may be observed within the epidermis, indicative of transepidermal elimination (192).
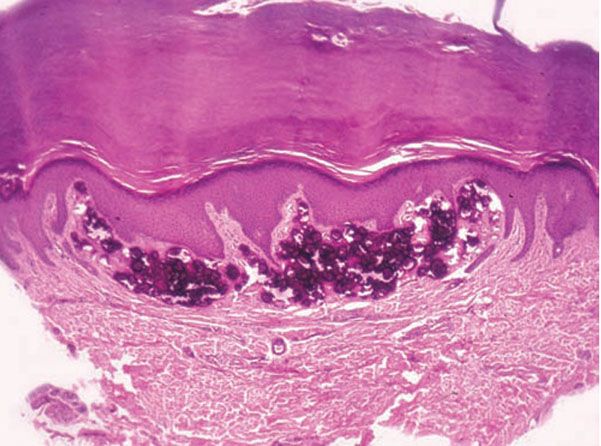
Figure 17-18 Subepidermal calcified nodule. A large deposit of calcium in the dermis distorts the overlying epidermis (H&E).
Pathogenesis. The primary event seems to be the formation of large, homogeneous masses that undergo calcification and break up into numerous calcified globules. Their origin of the homogeneous masses is obscure. It is not likely that they originate from a specific preexisting structure, such as sweat ducts or nevus cells, as has been assumed. Some suggest degranulation of mast cells with subsequent calcium and phosphate deposition as the etiology (195).
Principles of Management. Treatment is typically surgical excision.
GOUT
Clinical Summary. In the early stage of gout, there usually are irregularly recurring attacks of acute arthritis. In the late stage, deposits of monosodium urate form within and around various joints, leading to chronic arthritis with destruction of the joints and the adjoining bone. During this late stage, urate deposits, called tophi, may occur in the dermis and subcutaneous tissue. With improved treatments, the incidence of tophaceous lesions in gout significantly decreased from 1949 to 1972, from 14% to 3%, even though the incidence of gout remained unchanged. The prevalence and annual incidence of gout has increased over the last four decades, however, and updated studies to address the impact of these trends and the increased clinical complexity of the disease on the incidence of tophaceous gout are lacking (196). Tophaceous gout usually develops within 5 years of the initial onset of gout in 30% of untreated patients (197).
Cutaneous tophi are observed most commonly on the helix of the ears, olecranon bursae, and on the fingers and toes (197,198) (Fig. 17-19). They may attain a diameter of several centimeters. When large, tophi may discharge a chalky material (197). In rare instances, gout may present as tophi on the fingertips or as panniculitis on the legs without the coexistence of a gouty arthritis (199,200). Exceedingly rare instances of disseminated miliaral lesions have been reported (201,202).
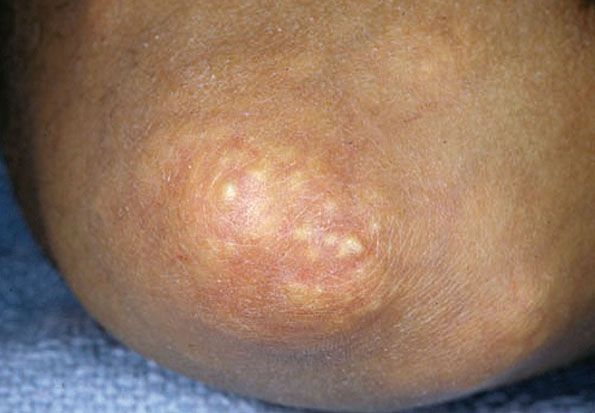
Figure 17-19 Gouty tophi. There are yellow-white urate deposits on the elbow.
Histopathology.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


