This chapter covers a diverse group of diseases that have in common a disturbance in metabolism or bodily function. Many of these conditions have a genetic basis, most of which have been elucidated in the last two decades.
Three major disease categories will be considered in this chapter:
• vitamin and dietary disturbances
• lysosomal storage diseases
• miscellaneous metabolic and systemic diseases.
The various diseases included in these categories show a wide range of histopathological changes. They will be discussed in turn.
VITAMIN AND DIETARY DISTURBANCES
The skin and mucous membranes may be affected in various vitamin deficiency states. Usually multiple vitamins are involved, as the most common cause of deficiency is a nutritional disturbance. Nutritional deficiencies may result from alcoholism, 1 digestive tract disease (including cystic fibrosis, 2 resections, and bypass surgery), dietary fads, and anorexia nervosa.3.4. and 5. The cutaneous manifestations of vitamin6 and nutritional7 deficiencies were reviewed some years ago. Cutaneous manifestations are particularly seen in vitamin C deficiency (scurvy), vitamin A deficiency (phrynoderma), and niacin (nicotinic acid) deficiency. These deficiencies are discussed further below. Vitamin D has little to do with cutaneous disease, but potential insufficiency of this vitamin as a consequence of sun protection methods will be discussed briefly below.
Kwashiorkor, which results from protein malnutrition, is characterized by xerosis, patches of hypopigmentation, skin peeling, peripheral edema, and thin hair shafts.7.8.9.10.11. and 12. It may follow a protein-poor diet.13. and 14. Deficiency of essential fatty acids, seen in some patients receiving parenteral nutrition, results in alopecia, xerosis, and intertriginous erosions. 7
Riboflavin and pyridoxine deficiency both lead to glossitis, angular stomatitis, cheilosis, and a condition resembling seborrheic dermatitis.7. and 15. In riboflavin deficiency there may be a scrotal dermatitis, while in pyridoxine deficiency there may be pellagra-like features. 7
Carotenemia, secondary to excessive dietary ingestion of foods containing β-carotene, is sometimes seen in young children fed commercial baby foods. 16 It presents with orange-tinted skin. It is not the only cause of xanthoderma. 17 Carotenemia can also be seen in adults with dietary fads. 18
SCURVY
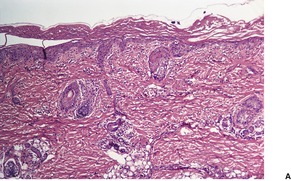
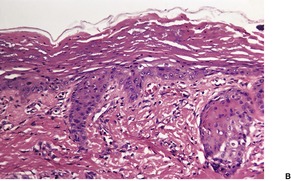
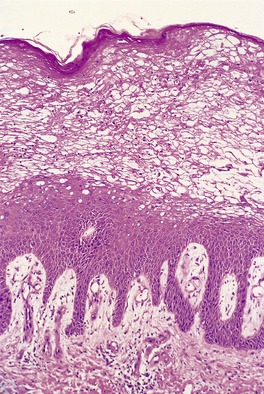
Scurvy results from a deficiency of vitamin C (ascorbic acid) which is a water-soluble vitamin necessary for proline hydroxylation in the formation of collagen.6.19. and 20. Vitamin C also plays a role in normal hair growth. Loss of the integrity of collagen leads to inadequate support for small vessels, resulting in hemorrhage from minor trauma. 21 This is characteristically perifollicular in distribution, but spontaneous petechiae and ecchymoses may also develop.6. and 22. Other features include follicular hyperkeratosis, abnormal hair growth with the formation of corkscrew hairs, 23 bleeding gums, and poor wound healing.6. and 24. Woody edema of the lower limbs with some surface scaling may be the only manifestation. 21 In one patient, the manifestations of scurvy were limited to a previously injured extremity. 25 Scurvy may be seen in alcoholics and those with dietary fads and inadequacies26.27.28. and 29. and, accordingly, associated deficiencies of other factors may contribute to the appearance of the cutaneous lesions. 30 It has also been seen in liver transplant recipients. 31 Dietary intake of vitamin C is essential, because it is not stored in the body. 32 Rarely, there is no explanation for the deficiency. 33
Histopathology
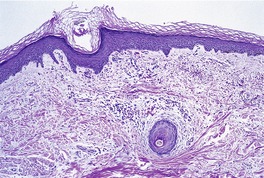
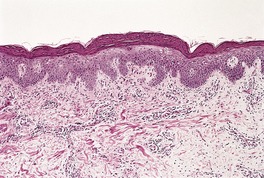
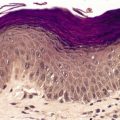
A characteristic feature is the presence of extravasated erythrocytes around vessels in the upper dermis (Fig. 18.1). This is often in a perifollicular distribution initially. Hemosiderin, a legacy of earlier hemorrhages, is sometimes found. There may also be follicular hyperkeratosis with coiled, fragmented, corkscrew-like hairs buried in the keratotic follicular material. 24 Ulceration of the skin is sometimes seen.
Fig. 18.1
Scurvy. The patient ate one brand of cookie as her only food. There is red cell extravasation, mild follicular hyperkeratosis, and perifollicular fibrosis. (H & E)
Electron microscopy
Affected skin may show alterations in fibroblasts, with defective collagen formation. 34 There may also be alterations in the endothelial cells of vessels and their junctions. 34
VITAMIN A DEFICIENCY
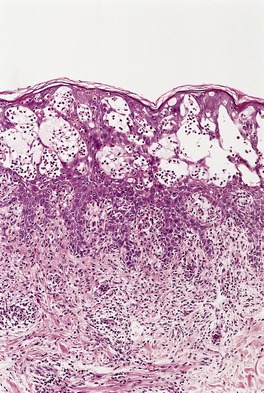
Vitamin A is a fat-soluble vitamin; its active form is retinol. Deficiencies are rare and usually related to malabsorption states.6.35.36. and 37. The skin becomes dry and scaly with follicular keratotic papules (phrynoderma). 38 Ocular changes include night blindness.39. and 40.
Histopathology
Sections show hyperkeratosis and prominent keratotic follicular plugging. 39 Sweat glands may be atrophic; in severe cases they show squamous metaplasia. 38
HYPERVITAMINOSIS A
Hypervitaminosis A is usually a result of self-administration of excess amounts of the vitamin.41.42. and 43. Acute symptoms include vomiting, diarrhea, and desquamation of skin. In the chronic form of vitamin A toxicity there is dry skin, cheilitis, and patchy alopecia. Histological changes are non-specific.
Vitamin K is a fat-soluble vitamin which is necessary for the hepatic synthesis or secretion of various coagulation factors. 6 A deficiency may result from liver disease and from malabsorption; in infants it may be associated with diarrhea. Purpura is a common manifestation of vitamin K deficiency.
The parenteral injection of vitamin K may rarely give rise to an erythematous plaque at the site of injection, apparently due to a delayed hypersensitivity reaction. 45 A late sclerodermatous reaction is a rare complication (see p. 313).
VITAMIN B12 DEFICIENCY
Vitamin B12 deficiency may be associated with poikilodermatous pigmentation. This clinical pattern results from basal pigmentation and some melanin incontinence. The nuclei of keratinocytes were reported to be larger than normal in one patient. 46
PELLAGRA
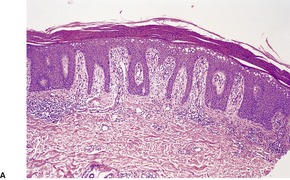
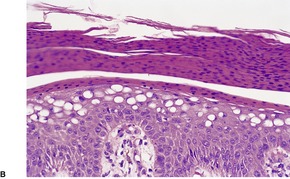
Pellagra is a multisystem nutritional disorder caused by inadequate amounts of niacin (nicotinic acid) in the tissues.47. and 48. This may result from a primary dietary deficiency,49.50.51.52.53. and 54. chronic alcoholism, 55 malabsorption, certain chemotherapeutic agents such as isoniazid,56. and 57. 6-mercaptopurine, 5-fluorouracil, 58 azathioprine, 59‘alternative remedies’, 60 anticonvulsants,61. and 62. and chloramphenicol, or from abnormalities of tryptophan metabolism. 6 In this latter category is the carcinoid syndrome, in which tumor cells divert tryptophan metabolism towards serotonin and away from nicotinic acid, and Hartnup disease, in which there is a congenital defect in tryptophan absorption and transfer6 (see p. 491). Nicotinamide, the active amide of niacin, is being investigated for its property of inhibiting photocarcinogenesis and photoimmunosuppression. 13
Pellagra is traditionally remembered as the ‘disease of the four Ds’: dermatitis, diarrhea, dementia and, if untreated, death. 63 The skin lesions commence as a burning erythema in sun-exposed areas, particularly the dorsum of the hands and the face and neck. 64 Blistering may occur. This is followed by intense hyperpigmentation with sharp margination and areas of epithelial desquamation. 6 The ‘tanning’ occurs more slowly than in typical sunburn. 65 There may also be glossitis, angular cheilitis, and vulvitis. 63
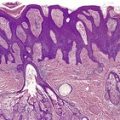
The findings are not diagnostic. They include hyperkeratosis, parakeratosis, epidermal atrophy with pallor of the upper epidermis and hyperpigmentation of the basal layer (Fig. 18.2).6. and 66. There is usually a mild, superficial dermal infiltrate of lymphocytes. Mild keratotic follicular plugging is sometimes seen in biopsies from the face. 56 Bullae may be either intraepidermal or subepidermal. 64 Hyperplasia of the sebaceous glands with follicular dilatation and plugging may occur. 47
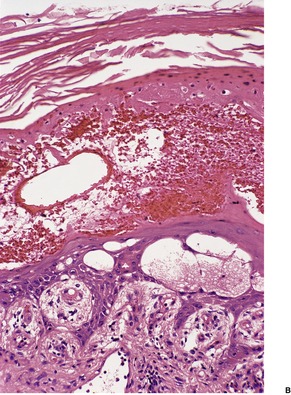
Fig. 18.2
(A) Pellagra. (B) There is partial necrosis and hemorrhage involving the superficial epidermis with underlying psoriasiform acanthosis. (H & E)
Lysosomal storage diseases are a group of individually rare, but collectively numerous, inherited disorders of intracellular metabolism. 67 More than 45 different disorders are recognized, many neurodegenerative in nature. Fifteen of these disorders account for 75% of lysosomal storage diseases. 67 They are a specific subset of the inborn errors of metabolism: they are characterized by a deficiency in a specific lysosomal hydrolase or of a protein essential for the normal function of lysosomes. 68 As a consequence of this deficiency there is accumulation of the specific substrate in various organs of the body. The distribution of this stored material corresponds to the site where degradation of the substrate usually occurs. Lysosomes are particularly plentiful in macrophages and other cells of the mononuclear phagocyte system: organs rich in these cells, such as the liver and spleen, are frequently enlarged. In one subgroup of lysosomal storage diseases, the sphingolipidoses, there is an accumulation of certain glycolipids or phospholipids in various organs, particularly the brain.
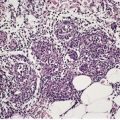
The lysosomal storage diseases can be diagnosed by assaying for the specific enzyme thought to be deficient in serum, leukocytes or cultured fibroblasts, 68 or the protein amount. 67 In many of these diseases inclusions can be found on ultrastructural examination of the skin (Fig. 18.3). The inclusions are sometimes sufficiently distinctive to be diagnostic of a particular disease, although in many instances they are not. Accordingly, ultrastructural examination of the skin in the diagnosis of the lysosomal storage diseases is usually no more than a useful adjunct to enzyme assay. 69 Recently a fluorescent analogue of lactosylceramide has shown promise as a screening test for the sphingolipidoses and some other lysosomal storage diseases. It accumulates in the lysosomes of cultured fibroblasts from affected patients. 70
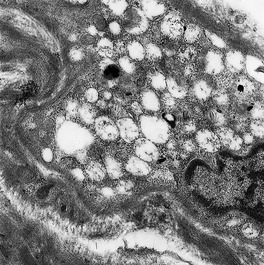
Fig. 18.3
Vacuolated fibroblasts in the skin in a lysosomal storage disease. The type of vacuole present is not diagnostic of a particular condition. (×5000)
With ultrastructural studies of the skin, care must be taken to avoid overdiagnosis. 69 Many cells in the skin may, at times, contain a few vacuoles, fat globules, or other inclusions. 69 These must not be misinterpreted as indicating a lysosomal storage disease.
The lysosomal storage diseases, which have a prevalence of 1 in 7700 in the general Australian population, can be divided into several categories on the basis of the biochemical nature of the accumulated substrate:71. and 72.
• sphingolipidoses
• oligosaccharidoses
• mucolipidoses
• mucopolysaccharidoses
• others.
SPHINGOLIPIDOSES
The sphingolipidoses are a heterogeneous group of lysosomal storage diseases that result from a variety of enzyme deficiencies affecting different levels in the metabolism of complex lipids. Certain glycolipids or phospholipids accumulate in various tissues of the body, particularly the brain. 73 Cutaneous changes are present in many of the sphingolipidoses. 69
GM2-gangliosidoses
The GM2-gangliosidoses are a subgroup of sphingolipidoses in which there is an accumulation of the ganglioside GM2, as a result of a defect in some aspect of the hexosaminidase system.68. and 74. The most common clinical variant is Tay–Sachs disease (OMIM 272800) in which there is progressive psychomotor deterioration and blindness. It is due to a mutation in the gene encoding the alpha subunit of the hexosaminidase A enzyme (HEXA) on chromosome 15q23–q24. In Sandhoff’s disease (OMIM 268800), which is phenotypically similar, the gangliosides are deposited in nearly all cells of the body, in contrast to Tay–Sachs disease in which deposits do not occur outside the nervous system.69. and 75. In Sandhoff’s disease the mutation involves the beta subunit of hexosaminidase (HEXB), on chromosome 5q13. 76
Histopathology
Cytoplasmic inclusions can be seen in a number of cells in osmificated, semithin, Epon-embedded sections in Sandhoff’s disease. 77
Electron microscopy
Membrane-bound inclusions can be found in endothelial cells, smooth muscle cells, pericytes, Schwann cells, and eccrine secretory cells. There are lamellar and vacuolar structures and ‘zebra bodies’ (vacuoles with transverse membranes). In Tay–Sachs disease, lesions are confined to nerve axons, which may be distended by residual bodies, a change also seen in Sandhoff’s disease. 69
Inclusion bodies are also found in cultured fibroblasts in Sandhoff’s disease, but they are quite sparse in Tay–Sachs disease. 78
GM1-gangliosidoses
GM1-gangliosidosis (OMIM 230500) is an autosomal recessive lysosomal storage disease caused by a mutation in the gene encoding β-galactosidase-1 (GLB1). There are two major clinical variants of GM1-gangliosidosis, of which Norman–Landing disease (pseudo-Hurler’s syndrome) is the more severe. 74 This infantile form (type I) is characterized clinically by a gargoyle-like appearance, psychomotor regression, blindness, hirsutism, hepatosplenomegaly, and deformities of the hands and feet.69. and 79. As in GM2 gangliosidosis, a ‘cherry-red spot’ is often present. Extensive dermal melanocytosis has been reported in several patients with GM1-gangliosidosis. 80
Vacuolation of fibroblasts, endothelial cells, and eccrine secretory cells is sometimes discernible in sections stained with hematoxylin and eosin. 79
In patients with associated dermal melanocytosis, lesional skin shows elongated melanocytes with fine melanin pigment. 80
Electron microscopy
There is vacuolation of fibroblasts, endothelial cells, smooth muscle cells, and sweat gland epithelium. Schwann cells are less severely affected. 69 The vacuoles are empty or contain fine fibrillar or flocculent material. 81 Inclusions are also found in cultured fibroblasts. 78
Gaucher’s disease
Gaucher’s disease is the most prevalent sphingolipidosis with a frequency of 1 in 50000–100000 live births in white populations, and 1 in 850 in Ashkenazi Jews. 82 Gaucher’s disease is categorized phenotypically into three main subtypes. All forms are caused by mutations in the gene encoding glucocerebrosidase (GBA) on chromosome 1q21; more than 200 mutations are known.83.84.85. and 86. Type I Gaucher’s disease (OMIM 230800) is the most common form of the disease; it lacks central nervous system involvement, as occurs in the other types. 87
In Gaucher’s disease, glucocerebroside accumulates in the cells of the mononuclear macrophage system (reticuloendothelial system).88.89. and 90. This condition will not be discussed further as skin biopsies have consistently been negative, with no evidence of stored lipid in sweat ducts, fibroblasts, or cutaneous nerves.69. and 81.
Fabry’s disease
Fabry’s disease (OMIM 301500) is an uncommon X-linked recessive disorder of glycosphingolipid metabolism in which there is a deficiency of the lysosomal hydrolase, α-galactosidase A (formerly called ceramide trihexosidase).91.92. and 93. The gene maps to Xq22. This leads to the accumulation of ceramide trihexoside in various tissues of the body, particularly the vascular and supporting elements.
The disease usually presents in late adolescence with recurrent fevers associated with pain in the fingers and toes and intermittent edema.91.94.95. and 96. Characteristic whorled opacities are usually present in the cornea (cornea verticillata).91. and 92. Cerebrovascular and cardiovascular disturbances are common, and progressive renal damage leading to renal failure in the fourth and fifth decades of life is almost invariable.94. and 97. Atopic eczema is not increased. 98 Heterozygous females are often asymptomatic, but they may show evidence of the disease to different degrees.99.100.101. and 102. Therefore, they are more than ‘carriers’ of the gene.
The diagnosis of Fabry’s disease can be made on the urine lipid profile, and by the reduced α-galactosidase A activity in peripheral blood leukocytes. 102
The cutaneous lesions (angiokeratoma corporis diffusum) are multiple, deep red telangiectases clustered on the lower part of the trunk, buttocks, thighs, scrotum, and the shaft of the penis (see p. 896). 91 Angiokeratomas were present in 66% of males and 36% of females registered on the Fabry Outcome Survey, a multicenter European database of 714 patients (at time of the review). 103 Most sites can be involved, although the face and scalp are usually spared. Petechial lesions are rarely present. 104 Cutaneous lesions are occasionally absent. 92 Multiple leg ulcers have also been described. 105 There may be anhidrosis.92. and 103.
It should be noted that angiokeratomas are not confined to Fabry’s disease. They may also occur in sialidosis, fucosidosis, adult-onset GM1-gangliosidosis, aspartylglycosaminuria, β-mannosidosis, α-N-acetylgalactosaminidase deficiency, galactosialidosis, and in an idiopathic form.
The effect of enzyme replacement therapy on the vasculopathy in Fabry’s disease appears to be minimal. 106 As this therapy is expensive, 107 it is important that a correct diagnosis, with biochemical confirmation, is made prior to therapy.108. and 109.
Histopathology
There are large and small thin-walled vessels in the upper dermis (see p. 896). The overlying epidermis is often thinned, with variable overlying hyperkeratosis. There are often acanthotic or elongated portions of rete ridge at the periphery of the lesions. 110 The vessels are angiectatic and not a new growth. Fibrin thrombi are sometimes present in the lumen. 111 There may be patchy vacuolization of the media of vessels in the deep dermis in both affected and normal skin. If frozen sections are examined, doubly refractile material may be seen in the vicinity of these vacuoles. 91 The material will also stain with Sudan black and the PAS stain. 112 The peroxidase-labeled lectins of Ricinus communis and Bandeiraea simplicifolia have been found to be strongly reactive with the material on frozen sections. 113 Fine PAS-positive granules are sometimes seen in the sweat glands. 111
In semithin sections, fine intracytoplasmic granules can be seen in eccrine glands, vessel walls, and fibroblasts in the dermis.
Diagnostic intracytoplasmic inclusions having a lamellar structure can be found in endothelial cells, pericytes, fibroblasts, myoepithelial cells of sweat glands, and macrophages. Involvement of eccrine secretory cells is uncommon92 and that of Schwann cells is rare. 77 The inclusions, which are sometimes membrane bound, may also be found in heterozygotes in skin biopsies112 and cultured fibroblasts. 78
Metachromatic leukodystrophy
Metachromatic leukodystrophy (OMIM 250100) results from a deficiency in the activity of arylsulfatase A and the accumulation of metachromatic sulfatides in the nervous system and certain other organs.68. and 74. Clinically, there is progressive psychomotor retardation. The gene maps to chromosome 22q13.31–qter.
Histopathology
Vacuolated cells are sometimes seen in the endoneurium of cutaneous nerves. Brown metachromatic material can be seen in these nerves after cresyl violet staining of frozen sections. 115
The Schwann cells of myelinated nerves contain so-called ‘tuff-stone’ or ‘herring bone’ inclusions which are membrane bound.69.77. and 116. Macrophages containing myelin breakdown products are found within the nerves. Inclusions with a concentric lamellar structure have been reported in cultured fibroblasts. 78
Krabbe’s disease
Krabbe’s disease (globoid cell leukodystrophy – OMIM 245200) is an autosomal recessive disease that results from a mutation in the galactosylceramidase gene on chromosome 14q31. As a consequence, there is a deficiency of the lysosomal enzyme galactocerebrosidase.68. and 117. There are progressive neurological symptoms, beginning usually in childhood, as a consequence of the apoptotic death of oligodendrocytes and Schwann cells resulting from the accumulation of psychosine. 117 Its incidence in the United States is 1 in 100000 live births. 117
Globoid cells with PAS-positive cytoplasm are found in the central nervous system, particularly in the white matter. 118 Cutaneous nerves appear normal on light microscopy.
Electron microscopy
Tubular and crystalloid inclusions have been reported in Schwann cells in cutaneous nerves,77. and 118. but not consistently. 81 Cultured fibroblasts do not contain specific inclusions. 78
Disseminated lipogranulomatosis (Farber’s disease – OMIM 228000) is a rare, autosomal recessive disorder of lipid metabolism in which there is a deficiency of acid ceramidase leading to an accumulation of ceramide and its degradation products.119. and 120. The defect maps to chromosome 8p22–p21.3. Several mutations have been described. The main clinical features usually appear at the age of 2–4 months and comprise progressive arthropathy, the development of subcutaneous, often periarticular nodules, hoarseness, irritability, and pulmonary failure. 121 The disease is progressive, death usually occurring in early childhood. 121
The diagnosis can be made by demonstrating a deficiency in ceramidase in cultured fibroblasts or in white blood cells. 122
There is extensive fibrosis of the reticular dermis and subcutis with collagen bundles of variable thickness traversing the nodules in various directions. Within the fibrotic areas are many histiocytes with distended, somewhat foamy cytoplasm.120. and 124. Some cells are multinucleate. A few lymphocytes and plasma cells are often present. Histochemical stains have given variable results, depending on whether paraffin or frozen sections have been used. The oil red O stain and Baker’s reaction for phospholipid may be positive. 123
There are characteristic curvilinear bodies (Farber bodies) within the cytoplasm of fibroblasts and occasionally of endothelial cells. They are also found within phagosomes of histiocytes at various stages of degradation. Banana-like bodies can be found within Schwann cells. ‘Zebra bodies’ (vacuoles with transverse membranes) may be seen in some endothelial cells. They represent gangliosides and may be found in other storage diseases. 122
Niemann–Pick disease
Niemann–Pick disease is a rare autosomal recessive disorder in which sphingomyelin accumulates in many organs due to a deficiency of sphingomyelinase. 125 It is characterized by progressive neurodegeneration. It is a heterogeneous entity with more than one enzyme defect involved.126.127.128. and 129. Several types have been described: type A (OMIM 257200), the predominant type, is due to a mutation in the sphingomyelin phosphodiesterase-1 gene (SMPD1) on chromosome 11p15.4–p15.1; type B (OMIM 607616), which is allelic to type A, has visceral involvement, but no neurological disease (types E and F are included here); type C1 (OMIM 257220) and type D are caused by mutations in the NPC1 gene on chromosome 18q11–q12; and type C2 (OMIM 607625) is due to a mutation in the NPC2 gene at 14q24.3. 130 The NPC genes appear to regulate intracellular trafficking of cholesterol and other lipids. The course is unremitting and death usually occurs in early childhood.
Cutaneous lesions have been reported in a small number of cases and include diffuse tan brown hyperpigmentation, indurated brown plaques, 131 facial papules, 132 xanthomas, 125 and juvenile xanthogranulomas.133. and 134. A nodular panniculitis has been described in a patient with type C disease. 135 A small number of cases have abnormalities in skin barrier function as a consequence of a marked reduction in sphingomyelin-derived ceramide. 136
Histopathology
It is now thought that the lesions reported clinically as juvenile xanthogranulomas in Niemann–Pick disease are xanthomas associated with the basic phospholipid abnormality.127. and 133. This view is based on the presence of cytoplasmic zebra bodies on electron microscopy of one case. 133 In the cutaneous lesions described there are large numbers of foamy histiocytes in the dermis admixed with a few lymphocytes. The foamy cells may have a pale brownish appearance in sections stained with hematoxylin and eosin. 132 The vacuoles may impart a mulberry appearance. The cytoplasmic lipids stain with oil red O and Sudan black; 125 they are metachromatic with toluidine blue. 132 Scattered multinucleate cells are present.
Electron microscopy
Cultured fibroblasts from patients with Niemann–Pick disease show characteristic membrane-bound myelin-like inclusions. 78‘Washed-out’ inclusions with a lamellar structure have been reported in endothelial cells and Schwann cells in the skin. 81
OLIGOSACCHARIDOSES
The oligosaccharidoses (glycoproteinoses) are characterized by excess urinary excretion of oligosaccharides as a consequence of a deficiency in one of the lysosomal enzymes responsible for the degradation of the oligosaccharide portion of glycoproteins. 68 The four disorders included in this subgroup of lysosomal storage diseases are sialidosis, fucosidosis, mannosidosis, and aspartyl-glycosaminuria. The cutaneous manifestations of this last condition have not been studied extensively and it will not be considered further.
Sialidosis
Sialidosis (mucolipidosis I, neuraminidase deficiency – OMIM 256550) results from a deficiency of neuraminidase (sialidase), 137 caused by mutations in the NEU1 gene on chromosome 6p21.3. Clinical features include coarse facies, ataxia, myoclonus, and a cherry-red spot in the macula. 137
Galactosialidosis (OMIM 256540) is a slowly progressive neurodegenerative disease, with similar phenotypic features, which results from the combined deficiency of neuraminidase (sialidase) and β-galactosidase.138. and 139. Several different mutations have been described in the PPGB gene that maps to 20q13.1. There is the lack of a 32 kDa ‘protective protein’ that is crucial for the biological activity of these two enzymes. Angiokeratomas have been reported in patients with this combined deficiency state.139. and 140.
Histopathology
Skin biopsies in sialidosis appear normal in sections stained with hematoxylin and eosin. In the combined deficiency, angiokeratomas may be present (see p. 896): the endothelium of these vessels is sometimes vacuolated. 139
Electron microscopy
Cultured fibroblasts from patients with sialidosis contain cytoplasmic vacuoles similar to those observed in various mucopolysaccharidoses and in mannosidosis. 141
In galactosialidosis, vacuoles are seen in the endothelium of vessels and also in fibroblasts, sweat gland epithelium, and the Schwann cells of non-myelinated nerves. The vacuoles are mostly empty, but some contain floccular material. 139 Lamellar inclusions also occur. Vacuolar and lamellar inclusions are present in cultured fibroblasts. 141
Fucosidosis
Fucosidosis (OMIM 230000) is a rare autosomal recessive disorder in which a deficiency of the lysosomal enzyme α-L-fucosidase leads to the accumulation of fucose-containing glycolipids and other substances in various tissues.142. and 143. The gene FUCA1, on chromosome 1p34, encodes this enzyme; more than 20 different mutations have been described. 144 About 100 cases have been reported worldwide. 144 There is early onset of psychomotor retardation and other neurological signs. Three clinical variants have been reported, but only type 3 is associated with cutaneous lesions. 145 These lesions are indistinguishable from the angiokeratomas seen in Fabry’s disease (see p. 485).144.146. and 147. Hypohidrosis, increased palmoplantar vascularity, and widespread telangiectasia may also be present. 148
Bone marrow transplantation is reported to be an effective treatment. 144 Enzyme replacement therapy or gene therapy is unlikely to become available in the near future. 144
Histopathology
The angiokeratomas are composed of dilated vessels in the papillary dermis (see p. 896). The endothelial cells of these and other dermal vessels are vacuolated and somewhat swollen, leading to narrowing of the lumen of some small dermal vessels. 146 The eccrine secretory coils are lined by uniformly vacuolated cells. 146
There are membrane-bound vacuoles containing fine granular material in endothelial cells, fibroblasts, melanocytes, histiocytes, eccrine glands, and occasional pericytes. Lamellated bodies representing complex lipids are present in myoepithelial cells of sweat glands and in Schwann cells. 146 Both types of cytosome are sparsely distributed in epidermal keratinocytes.146. and 150. Smooth muscle cells are uninvolved. 146
Mannosidosis
Two types of mannosidosis have been identified: α-mannosidosis (OMIM 248500), caused by a mutation in the gene MAN2B1, found at 19cen–q12, encoding the lysosomal enzyme α-mannosidase; and β-mannosidosis (OMIM 248510), an extremely rare condition, due to a mutation in the MANBA gene, encoding β-mannosidase, on chromosome 4q22–q25.151. and 152.
In α-mannosidosis, the deficiency of the lysosomal enzyme α-mannosidase leads to an accumulation of mannose-containing oligosaccharides in various tissues of the body, including the nervous system. 153 Patients have a gargoyle-like facies and mental retardation. The condition runs a relatively benign clinical course.
In β-mannosidosis, there is facial dysmorphism, skeletal deformities, respiratory and skin infections, angiokeratomas, hepatosplenomegaly, and neurological abnormalities.151. and 152.
Histopathology
Biopsies from hyperplastic gingiva in α-mannosidosis have shown vacuolated histiocytes in the lamina propria containing PAS-positive material. 153 The angiokeratomas found in β-mannosidosis resemble those seen in other conditions.
Electron microscopy
Membrane-bound vacuoles containing fine granular material are present in many different cells of the body, including cultures of skin fibroblasts, in both forms of the disease.141. and 151.
MUCOLIPIDOSES
The mucolipidoses are a group of lysosomal storage diseases that have clinical and biochemical features of both the mucopolysaccharidoses and the sphingolipidoses. 68 Glycolipids and glycosaminoglycans accumulate in the tissues. Type I mucolipidosis has been reclassified as sialidosis (see above), while type IV (OMIM 252650), which results from a mutation in the gene (MCOLN1) encoding mucolipin-1, is an autosomal recessive neurodegenerative disease. The most widely investigated of the mucolipidoses is I-cell disease (mucolipidosis II – OMIM 252500), so named because of the numerous inclusions seen in fibroblasts cultured from patients with the disease. 68 Type III mucolipidosis (OMIM 252600) is regarded as a milder form of I-cell disease. They are allelic, but other subtypes of type III involving other genes have been recorded. 154
I-cell disease
I-cell disease (mucolipidosis II) is caused by a mutation in the GNPTAB gene. 155 I-cell disease (OMIM 252500) is an autosomal recessive neurodegenerative disorder, characterized by a marked intracellular deficiency of a number of lysosomal hydrolases and by a significant elevation of these enzymes in plasma. 156 It is caused by a deficiency of uridine-diphosphate-N-acetylglucosamine 1-phosphotransferase, the enzyme that phosphorylates mannose residues of glycoproteins to allow their delivery to lysosomes.157. and 158. The gene maps to chromosome 12q23.3, although there has been conflicting assignment to 4q21–q23.
Clinically, there is short stature, facial dysmorphism, progressive mental and motor retardation, and bony deformities.159. and 160. The skin is generally pale and smooth. 159
Histopathology
The dermis may have an increased number of oval or spindle-shaped cells, some with clear or foamy cytoplasm. 159 The cytoplasmic inclusions are PAS positive and metachromatic; they stain with oil red O in frozen sections.
Electron microscopy
There are membrane-bound vacuoles in the cytoplasm of various cells, including fibroblasts, pericytes, Schwann cells, secretory cells of the eccrine glands, and endothelial cells.69. and 160. The vacuoles may contain a few dark rings. The inclusions in endothelial cells are more electron dense and multivesicular. 159 Cultured fibroblasts contain electron-dense inclusions. 141
In mucolipidosis IV, there are small dense lipid and zebra bodies in addition to the vacuoles. 69
OTHER LYSOSOMAL STORAGE DISEASES
Included in this group are glycogenosis type II, the neuronal ceroid-lipofuscinoses, a heterogeneous group of neurodegenerative disorders, and cystinosis.
The mucopolysaccharidoses are another major group of lysosomal storage diseases. They are considered with the mucinoses in Chapter 13 (see p. 367).
Trimethylaminuria (OMIM 602079), also known as fish-odor syndrome, is a rare metabolic disorder, characterized by a body malodor similar to that of decaying fish. 161 The condition is caused by mutations in the flavin-containing monoxygenase 3 (FMO3) gene, on chromosome 1q23–q25, resulting in the accumulation of trimethylamine. 161
Glycogenosis (type II), also known as glycogen storage disease II (OMIM 232300), is an autosomal recessive disease, caused by mutations in the gene (GAA) encoding acid alpha-1, 4-glucosidase (acid maltase), which maps to chromosome 17q25.2–q25.3. 68 In the classic infantile form (Pompe’s disease) children are hypotonic with enlarged hearts.
Histopathology
Glycogen is present in many cells in the skin and is best seen in biopsies fixed in Carnoy’s solution and stained by the PAS method.
Electron microscopy
Clustered glycogen granules, enclosed within a limiting membrane, are found in many types of cell in the skin, including the arrector pili muscles.69.77. and 81.
Neuronal ceroid-lipofuscinoses
The neuronal ceroid-lipofuscinoses are a heterogeneous group of progressive neurodegenerative disorders characterized by the accumulation of autofluorescent ceroid or lipofuscin-like substances in various organs, especially the nervous system.162.163. and 164. Types 1 to 10 have been identified, all with different genetic abnormalities and gene localizations. They were originally identified by the age of onset, but this has been discontinued since the genetic abnormalities have been identified. Type 1 (OMIM 256730) was the original infantile form. Abnormal peroxidation of fatty acids may be the metabolic basis. 162
Histopathology
Affected cells contain yellow-brown pigment which is autofluorescent.77. and 162.
Characteristic membrane-bound inclusions with a curvilinear or finger-print pattern have been reported in eccrine secretory cells, endothelial cells, smooth muscle cells, and macrophages. 69 They are particularly prominent in endothelial cells. 81 Inclusions have not been found consistently in fibroblasts and Schwann cells. 166 In one report only granular osmiophilic deposits were found; there were no curvilinear or finger-print inclusions in the cytoplasm of several cell types in the dermis. 167
Cystinosis
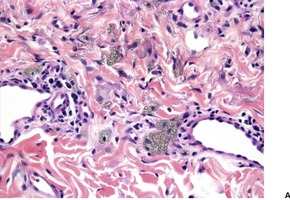
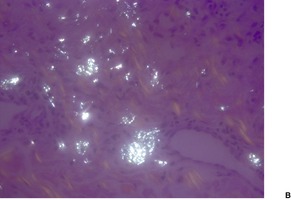
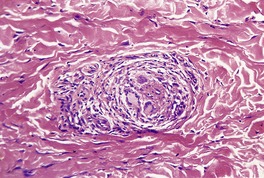
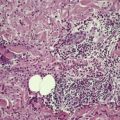
Cystinosis (OMIM 219800) is a lysosomal storage disease resulting from an error in the transmembrane transport of the amino acid cystine. It results from a mutation in the gene encoding cystinosin on chromosome 17p13. There is storage of cystine in multiple organs of the body, particularly the kidney, pancreas, cornea, central nervous system, and muscle. Skin and hair pigmentation is reduced, possibly resulting from impaired pigment formation in melanosomes. Cystine deposits in macrophages in the skin (Fig. 18.4).
Fig. 18.4
Cystinosis. (A) The brownish crystals are in macrophages. There is perivascular accentuation. (H & E) (B) The crystals are doubly refractile under polarized light. (Slides by courtesy of Dr Mark Wilsher, DHM, Sydney)
NECROLYTIC ERYTHEMAS
The necrolytic erythemas are a group of cutaneous diseases of diverse metabolic origin but which share certain histopathological features. In addition to the major conditions of acrodermatitis enteropathica, glucagonoma syndrome, necrolytic acral erythema, and Hartnup disease, several other rare conditions are discussed very briefly because of some shared features with these prototype conditions.
ACRODERMATITIS ENTEROPATHICA
Acrodermatitis enteropathica (OMIM 201100) is a rare autosomal recessive defect in the zinc transporter protein ZIP4, encoded by the SLC39A4 (hZIP4) gene on chromosome 8q24.3.168.169. and 170. Additional zinc transporters may be involved in this condition.169. and 171. It has an estimated incidence of 1 per 500000 children. 171 It usually presents in infancy, at the time of weaning, with the triad of alopecia, diarrhea, and dermatitis.172. and 173. The cutaneous lesions are periorificial and acral in distribution. There is a crusted eczematous eruption which is sometimes vesiculobullous or pustular. 174 Intercurrent infection with bacteria and yeasts, possibly related to impaired chemotaxis, 175 complicates the clinical picture.174. and 176. Other features include photophobia, nail dystrophy, hair shaft abnormalities, 177 short stature, progressive depigmentation, 178 stomatitis, and emotional disturbances. There are uncommon, mild forms of the disease, 179 some of which may not be diagnosed until adult life.180. and 181.
Transient symptomatic zinc deficiency may also develop in advanced cancer182 and in premature infants on artificial feeding, 183 and rarely in breastfed infants, both premature169.170.184.185.186.187. and 188. and full-term,189. and 190. associated with low or marginal levels of zinc in maternal milk. 191 Premature infants are more vulnerable to the development of zinc deficiency than full-term infants because they have low body stores of zinc and a poor capability to absorb zinc from the gut, despite their high zinc requirements.192. and 193. Sometimes serum zinc levels may be misleadingly normal. 194 Other rare causes of an acrodermatitis enteropathica-like eruption have included parenteral nutrition without zinc supplementation,195.196. and 197. malnutrition, 198 Crohn’s disease, 199 intestinal bypass procedures, 200 gastrectomy, 201 advanced alcoholic cirrhosis, 202 the nephrotic syndrome, 203 low dietary zinc in adults, 204 the acquired immunodeficiency syndrome, 205 Hartnup disease, 206 arginine deficiency, 207 ornithine transcarbamylase deficiency (OMIM 311250),207.208. and 209. an X-linked disease on chromosome Xp21.1, isoleucine deficiency, 210 anorexia nervosa,211. and 212. and cystic fibrosis.213.214. and 215.
The abnormality in acrodermatitis enteropathica, referred to above, involves a novel zinc transporter protein belonging to the ZIP (zinc/iron-regulated transporter-like protein) family. 168 Acrodermatitis enteropathica is responsive to zinc therapy;216. and 217. it needs to be life long. 218 Several different factors may be implicated in transient symptomatic zinc deficiency. These include diminished tissue stores of zinc in premature infants, the decreased bioavailability of zinc in cow’s milk when compared to human breast milk, and the rare, idiopathic occurrence of low zinc levels in breast milk, despite normal serum levels. 184
A periorificial dermatitis resembling acrodermatitis enteropathica may occur in the rare aminoacidopathies, methylmalonic (OMIM 251000) and propionic acidemia (OMIM 606054).219.220. and 221. An ichthyosis vulgaris-like rash has also been reported in methylmalonic acidemia. 222 The histopathological changes have been variable.
Selenium deficiency, an essential trace element like zinc, may produce a cardiomyopathy and cutaneous changes similar to zinc deficiency. 223 It may follow parenteral nutrition of long duration. 223
Biotin deficiency can also mimic zinc deficiency clinically. 224 It can result from acquired deficiencies (such as from the consumption of raw egg whites) or from an inborn error in metabolism, such as biotinidase or multiple carboxylase deficiency.225. and 226. Holocarboxylase synthetase deficiency (OMIM 253270) is a rare autosomal recessive disorder of the biotin metabolism that presents at birth with an erythrodermic eruption of the eyebrows and scalp. It may also present as an ichthyosis. 227 The gene, HLCS, is on chromosome 21q22.1.
Necrolytic erythema of the skin was seen in a patient with the exceedingly rare deficiency in glutamine synthetase (OMIM 610015), encoded by the GS (GLUL) gene on 1q31. 228 It is involved in ammonia detoxification. Glutamine is largely absent from serum, urine, and cerebrospinal fluid. 228
An amicrobial pustulosis of the flexures and scalp has been reported in association with various autoimmune diseases. 229 It is mentioned here because of its response to zinc supplementation. Another recently recognized aseptic condition has been called aseptic abscesses. Visceral organs are involved. The lesions respond to corticosteroids, but not antibiotics. 230 About two-thirds of affected patients have inflammatory bowel disease. 230
The histological changes, which vary with the age of the lesion, are similar to those seen in the necrolytic migratory erythema of the glucagonoma syndrome (see below). In early lesions, there is confluent parakeratosis overlying a normal basket-weave stratum corneum. 231 The granular layer is absent and there is mild spongiosis and acanthosis. There is increasing pallor of the cells in the upper layers of the epidermis and variable psoriasiform epidermal hyperplasia. 232 Subcorneal or intraepidermal clefts may develop but established vesiculobullous lesions are intraepidermal in location and result from cytoplasmic vacuolar change with massive ballooning and reticular change producing cytolysis of keratinocytes.233. and 234. Confluent necrosis leads to enlargement of the vesicles. Sometimes there is necrosis of the upper epidermis, but this was not encountered in one detailed study. 235
In late lesions, there is confluent parakeratosis overlying psoriasiform epidermal hyperplasia, but there is no significant epidermal pallor (Fig. 18.5). Less common findings include apoptotic cells, 236 a few acantholytic cells in vesiculobullous lesions, 237 and neutrophils within the epidermis. Secondary infection may complicate the picture.
Fig. 18.5
(A) Acrodermatitis enteropathica. (B) This late lesion is characterized by confluent parakeratosis overlying an acanthotic epidermis. (H & E)
Blood vessels in the papillary dermis are often dilated and there is a mild perivascular infiltrate of chronic inflammatory cells.
Pallor of the epidermal cells is also seen in the exceedingly rare deficiency of the M-subunit of lactate dehydrogenase (OMIM 150000), reported from Japan and mapped to 11p15.4;238. and 239. a similar disorder has been reported from Europe as ‘annually recurring acroerythema’. 240
Electron microscopy
Findings include lipid droplets and multiple cytoplasmic vacuoles in keratinocytes in the upper dermis.195.231. and 241. Desmosomes may be diminished, associated often with widening of the intercellular space. 231
GLUCAGONOMA SYNDROME
The clinical features of the rare glucagonoma syndrome include a distinctive cutaneous eruption (necrolytic migratory erythema), glossitis, stomatitis, diabetic type of glucose intolerance, scotoma, brittle nails, 242 anemia, weight loss, venous thrombosis, elevated glucagon levels, and decreased plasma amino acids.243.244.245. and 246. A glucagon-secreting islet cell tumor of the pancreas is usually present and is malignant in the majority of cases.243.247.248.249. and 250. The syndrome has also been reported in association with a jejunal adenocarcinoma, 251 in pancreatic insufficiency, 252 in association with a neuroendocrine tumor producing predominantly insulin, 253 in advanced cirrhosis of the liver,254. and 255. in association with villous atrophy of the small intestine, 252 in malnutrition, 256 after intravenous glucagon for hypoglycemia resulting from an insulin-like tumor product, 257 and in a patient with elevated glucagon levels but no detectable tumor. 258 Impairment of hepatic function has been present in many of the cases of necrolytic migratory erythema without a glucagonoma.259.260. and 261. Zinc deficiency is sometimes present as well in patients with cirrhosis of the liver,261. and 262. particularly if alcohol related, and combined with malnutrition. 263
The cutaneous lesions, called necrolytic migratory erythema because of their similarities to both toxic epidermal necrolysis and annular erythema, are manifest by waves of extending annular or circinate erythema and superficial epidermal necrosis with shedding of the skin leading to flaccid bullae and crusted erosions. 264 There is usually complete resolution of involved areas within 10–14 days.265. and 266. The lesions primarily affect the trunk, groin, perineum, thighs and buttocks, but the legs, perioral skin, 266 and sites of minor trauma may also be involved. Cutaneous lesions are not invariably present in the syndrome. Rarely, they are its only manifestation.267. and 268. Necrolytic migratory erythema has been reported in two patients with opiate dependency. In one case, the rash settled on withdrawal of methadone, only to return on recommencing methadone. 269 It has also been reported in a patient with cystic fibrosis. 270
The pathogenesis of the skin lesions is uncertain, but their histological similarities to those seen in other deficiency states, such as pellagra and acrodermatitis enteropathica, and their disappearance with intravenous administration of supplemental amino acids suggest that profound amino acid deficiency induced by the catabolic effects of hyperglucagonemia may be important.266. and 271. Elevated levels of arachidonic acid, an inflammatory mediator, have been found in affected skin. 272
Lanreotide, an analogue of somatostatin, but with a much longer half-life, has been used to treat the glucagonoma syndrome. It does not appear to suppress tumor growth. 273 Liver transplantation was used to treat a patient with metastatic glucagonoma. 274
Several histological patterns may be seen in necrolytic migratory erythema, depending on the stage of evolution of the lesion that is biopsied (Fig. 18.6). The most distinctive pattern is the presence of pale, vacuolated keratinocytes in the upper epidermis, leading to focal or confluent necrosis (Fig. 18.7). 258 This process has been termed ‘necrolysis’. 265 Subcorneal or intraepidermal clefts may result; acantholytic cells are rarely found in these clefts. 276 Subcorneal pustules are sometimes found adjacent to the areas of necrosis, but they may also be the only manifestation of disease in the biopsy specimen. 275 Diffuse neutrophilic infiltration of the epidermis may accompany this pattern (Fig. 18.8). 277
Fig. 18.6
Glucagonoma syndrome. This is an established lesion with a thick zone of pale, vacuolated keratinocytes in the upper epidermis. (H & E)
Fig. 18.7
(A) Glucagonoma syndrome with psoriasiform hyperplasia. (B) Vacuolated cells are quite conspicuous in the upper layers of the epidermis. (H & E)
Fig. 18.8
Glucagonoma syndrome. A pustular lesion is present. (H & E)
The least common histological pattern is psoriasiform hyperplasia of the epidermis with overlying confluent parakeratosis and vascular dilatation with some angioplasia in the papillary dermis (Fig. 18.9).258. and 278.
Fig. 18.9
Glucagonoma syndrome. Confluent parakeratosis overlies an epidermis in which there is mild psoriasiform hyperplasia. (H & E)
In all biopsies there is usually a mild to moderate perivascular infiltrate of lymphocytes in the upper dermis. Sometimes there are occasional neutrophils as well, particularly if subcorneal pustules are present.
Uncommon histological findings include a suppurative folliculitis, the presence of concomitant candidosis, 279 suprabasal acantholysis, 280 and scattered dyskeratotic cells in the upper epidermis. 281 In one series of 13 patients, the biopsy findings were regarded as suggestive or characteristic of necrolytic migratory erythema in only eight. 282 The author has seen similar cases, in which several biopsies were performed, but only one showed diagnostic features.
Electron microscopy
In one study, there was widening of the intercellular spaces in the upper epidermis and a reduction in the number of desmosomes. 283 The cytoplasm of affected cells showed vacuolar degeneration with lysis or absence of organelles. 283 Scattered dyskeratotic cells were noted.
NECROLYTIC ACRAL ERYTHEMA
Necrolytic acral erythema, first described in 1996, belongs to the family of necrolytic erythemas. 284 This variant is unique in its acral location and its strong association with hepatitis C.285.286.287. and 288. Clinically, there are eroded erythematous to violaceous patches and tender, flaccid blisters and erosions with hyperkeratotic plaques in older lesions. There is a predilection for the lower limbs. The condition responds to treatment with interferon alfa and oral zinc, although zinc levels were normal in all the earlier reports.285. and 289. Recently, zinc deficiency has been reported in rare patients with this condition.290.291. and 292. Combination therapy with interferon, ribavirin, and hyperalimentation has also been used with success. 293
The lesions resemble acrodermatitis enteropathica and necrolytic migratory erythema with hyperkeratosis, parakeratosis, superficial pallor of the epidermis, focal necrosis, and spongiosis. There is a superficial mixed inflammatory cell infiltrate in the dermis. 285
A histologically similar process has been reported in two patients with anorexia nervosa, following nutritional infusions. 294 Clinically the lesions were linear and striae-like.294. and 295.
HARTNUP DISEASE
Hartnup disease (OMIM 234500), named after the first family to be described with this condition, 296 results from defective intestinal absorption of tryptophan and impaired renal tubular reabsorption of neutral amino acids. 297 It is caused by mutations in the solute carrier family 6, member 19 (SLC6A 19) gene on chromosome 5p15. 298 There is a photosensitive, pellagra-like skin rash, cerebellar ataxia, mental disturbances, aminoaciduria, and indicanuria. 299 Symptoms commence in childhood; there is often some improvement in later life. Other genetic defects of tryptophan metabolism, resulting in some symptoms in common with Hartnup disease, have been described. 300 An acrodermatitis enteropathica-like eruption has also been reported in a child with Hartnup disease, 206 the reason for its inclusion here.
The changes in the skin are similar to those seen in pellagra (see p. 483).
MISCELLANEOUS METABOLIC AND SYSTEMIC DISEASES
As the heading suggests, this section deals with a heterogeneous group of disorders with variable clinical and histopathological manifestations. The various cutaneous manifestations of celiac disease are considered in other sections. They include xerosis, 301 dermatitis herpetiformis, alopecia areata, chronic urticaria, vasculitis, Sjögren’s syndrome, lupus erythematosus, and linear IgA dermatosis. 302
PROLIDASE DEFICIENCY
Prolidase deficiency (peptidase D – OMIM 170100) is an exceedingly rare, autosomal recessive, inborn error of metabolism in which recalcitrant leg ulcers are the most characteristic feature.303.304.305.306.307.308. and 309. It is caused by a mutation in the peptidase D (PEPD) gene which maps to 19cen–q13.11. 306 Prolidase splits iminodipeptides found in collagen. Other clinical features that may be present include mental retardation, splenomegaly, recurrent infections, a characteristic facies, and premature graying of the hair.310.311.312. and 313. Telangiectasia, photosensitivity, lymphedema, and erosive cystitis304 are rare manifestations. 303 The development of a squamous cell carcinoma in an ulcer is a surprisingly uncommon complication. 314 Onset of symptoms occurs in childhood. Large amounts of iminodipeptides are present in the urine. 315
Apheresis exchange has produced improvement in the leg ulcers in this condition. 316
Histopathology
The cutaneous ulcers may show secondary infection and variable fibrosis in chronic cases. Two reports have mentioned the presence of amyloid-like material in vessel walls and in the immediately adjacent dermis.317. and 318. Vascular wall thickening and infiltration of mononuclear cells and neutrophils have been observed in indurated lesions prior to their ulceration. 319 Although the dermal collagen in non-ulcerated areas appears normal on light microscopy, the fibers are seen to be smaller and irregularly patterned312 on electron microscopy. Elastic fibers are fragmented. 312
TANGIER DISEASE
Tangier disease (OMIM 205400) is a rare autosomal recessive disorder of plasma lipid transport in which there is a deficiency of normal high-density lipoproteins (HDL) in the plasma and an accumulation of cholesterol esters in many organs, particularly in the reticuloendothelial system. 320 The disorder was originally described in a kindred living on Tangier Island in Chesapeake Bay. The presence of enlarged yellowish tonsils and a low plasma cholesterol level is pathognomonic. The skin usually appears clinically normal, although small papular lesions have been described. 321 The disorder is caused by a mutation in the ATP-binding cassette-1 gene (ABC1). Abnormalities in this gene also cause another disease characterized by HDL deficiency. That is, the two conditions are allelic. The gene responsible maps to chromosome 9q22–q31.
Histopathology
Biopsies from clinically normal skin show perivascular and interstitial nests of foam cells admixed with a few lymphocytes and plasma cells. 322 In frozen sections, the cytoplasm of the foam cells stains with oil red O and Sudan black. 322 Doubly refractile cholesterol esters are demonstrable in both an intracellular and extracellular location. 322 In semithin sections there is extensive vacuolization of the cytoplasm of Schwann cells in small, unmyelinated cutaneous nerves. 320
Electron microscopy
The deposits are electron lucent and vary from spherical to crystalline in shape. They are not membrane bound. Lipid deposits are present in the cytoplasm of Schwann cells. 320
LAFORA DISEASE
Lafora disease (myoclonic epilepsy – OMIM 254780) is a familial, degenerative disorder with the clinical triad of seizures, myoclonus, and dementia.323. and 324. Cutaneous lesions are rarely present. 323 Lafora disease can be caused by mutations in the laforin (EPM2A) or the malin (NHLRC1) gene. Malin appears to regulate laforin protein concentrations. 325 The genes responsible map to chromosome 6q23–q25. 326 The OMIM gene map locus is listed as 6q24, 6p22.3. The intracytoplasmic inclusion bodies found in various organs, particularly in ganglion cells in parts of the brain, are glucose polymers called polyglucosans;323. and 327. they were first described by Lafora, who considered them to consist of amyloid.
Histopathology
The inclusion bodies (Lafora bodies, polyglucosan bodies) are well seen in the excretory ducts of eccrine and apocrine sweat glands of clinically normal skin.324.328. and 329. They are PAS positive and diastase resistant. The number of inclusions may vary with the biopsy site; 323 axillary skin is favored. 326
Electron microscopy
The inclusions are round or oval, non-membrane bound and often juxtanuclear in position.116. and 324. They are composed of fine filamentous material, dark-staining granules and vacuoles. 323
ULCERATIVE COLITIS AND CROHN’S DISEASE
Ulcerative colitis and Crohn’s disease (regional enteritis) have many cutaneous manifestations in common.330.331.332.333. and 334. Skin lesions occur in 10–20% of patients with either disease, but the incidence varies widely from one study to another, depending on the inclusion or otherwise of oral, perianal, and non-specific lesions.330. and 331. In the case of Crohn’s disease, cutaneous manifestations are more common in patients with colonic rather than ileal disease. The onset of skin lesions occasionally precedes the symptoms and signs of the inflammatory bowel disease. There is no correlation, as a rule, with the severity or activity of the bowel disease.
Erythema nodosum and pyoderma gangrenosum are the most common and specific cutaneous manifestations of both diseases, although pyoderma gangrenosum is more frequent in ulcerative colitis.335.336. and 337. Finger clubbing, aphthous ulcers of the mouth, 338 cutaneous polyarteritis nodosa,339. and 340. psoriasis, 341 pyostomatitis vegetans,342.343. and 344. erythema multiforme, 345 and vitiligo346. and 347. have been reported in both conditions. 330 Inflammatory bowel disease was present in 21 of 29 patients with aseptic abscesses, characterized by deep, sterile abscesses involving particularly the spleen, liver, and lymph nodes. 230 A small number of patients have a neutrophilic dermatosis. 230 Cutaneous complications of therapy sometimes develop.
In Crohn’s disease, the perianal manifestations include skin tags, fistulas, and abscesses.348. and 349. They are found in up to 80% of individuals with colonic involvement. 350 Mucosal ‘cobblestoning’ and fissuring may occur in the mouth.351.352. and 353. Intraepithelial IgA pustulosis is a rare oral disease occurring in Crohn’s disease. 354 Cheilitis granulomatosa (see p. 188) may occur on the lips. 355 Other lesions described in Crohn’s disease include erythema elevatum diutinum, 356 a vesiculopustular eruption, 357 a neutrophilic dermatosis of the malar regions, 358 epidermolysis bullosa acquisita, 359 acne fulminans, 360 pyoderma faciale, 361 a neutrophilic lobular panniculitis, 362 granuloma annulare/necrobiosis lipoidica-like lesions, 337 granulomatous vasculitis (Fig. 18.10),337. and 363. porokeratosis, and nutritional deficiency states related to zinc, niacin (nicotinic acid) and vitamin C.330. and 351. The occurrence of granulomas in nodular, ulcerated or plaque-like lesions, at sites well removed from involved mucosal surfaces, has been called ‘metastatic Crohn’s disease’.364.365.366.367.368.369.370.371.372.373.374. and 375. Some lesions are in intertriginous areas; the limbs are another favored site. 376 The vulva, breast, 377 scrotum, and penis are rarely involved.378.379.380.381.382.383. and 384. Genital involvement appears to be more common in children than adults.385.386.387. and 388. There are non-caseating granulomas, similar to those seen in the bowel, scattered through the dermis and sometimes the subcutis. Granulomas with a slight cuff of lymphocytes are present in a nodular or diffuse pattern with an associated superficial and deep perivascular mixed inflammatory infiltrate. 389 Eosinophils are sometimes conspicuous. 389 Sometimes there are only occasional granulomas in a perivascular distribution (granulomatous perivasculitis). 390
Fig. 18.10
Crohn’s disease. A small, non-caseating granuloma is in intimate contact with a small blood vessel in the lower dermis. There is extravasation of fibrin into the vessel wall. (H & E)
Uncommon manifestations of ulcerative colitis include thromboembolic phenomena, cutaneous vasculitis,335. and 391. and a vesiculopustular eruption. 392 Some of the pustular lesions described in ulcerative colitis probably represent evolving lesions of pyoderma gangrenosum, others resemble Sweet’s syndrome,393. and 394. while still others are non-specific pustular eruptions.391.395.396.397.398. and 399. These may show either a suppurative folliculitis or intraepidermal or deeper abscesses. In short, a spectrum of neutrophilic dermatoses can occur. 393 Granulomas with admixed neutrophils were present in one proven case of ulcerative colitis. 400
WHIPPLE’S DISEASE
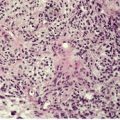
Whipple’s disease is a rare, multisystem, bacterial infection characterized by malabsorption, abdominal pain, arthritis, and neurological manifestations. The organism responsible, Tropheryma whippelii, is related to the actinomycetes group of Gram-positive bacteria. 401 The cutaneous changes include hyperpigmentation of scars and sun-exposed skin, observed in approximately 40% of patients, 402 as well as erythema nodosum and, rarely, subcutaneous nodules.403.404. and 405.
The subcutaneous nodules show a non-specific panniculitis with pockets of foamy macrophages containing PAS-positive, diastase-resistant material and resembling those seen in small bowel biopsies. 405
CYSTIC FIBROSIS
Cystic fibrosis (OMIM 219700) is an autosomal recessive disorder due to mutations in the cystic fibrosis conductance regulator (CFTR) gene. It occurs in 1 in 2500 live births in white populations. 407 Cutaneous manifestations of malnutrition are seen infrequently in cystic fibrosis; they have been attributed to deficiencies of protein, zinc, and fatty acids.2.213.408. and 409. The lesions described include erythematous, desquamating papules and plaques, and scaling, sometimes annular, patches and plaques. 408 Aquagenic skin wrinkling may develop at an early stage. 407 An acrodermatitis enteropathica-like eruption has also been described.214. and 215. In another patient the eruption resembled necrolytic migratory erythema. 270 The lesions develop early in life.
The changes are not diagnostically specific. They include acanthosis, a diminished granular layer with overlying parakeratosis, and a mild perivascular infiltrate of lymphocytes in the upper dermis. Mild spongiosis is sometimes present. The necrolysis and pallor seen in acrodermatitis enteropathica are absent.
DIABETES MELLITUS
Cutaneous manifestations are common in diabetics, occurring at some time in approximately 30% of all people who have the disease. Most of these skin complications and associations have been discussed elsewhere in this volume, but they are listed below for completeness. Three complications that have not been considered elsewhere – microangiopathic changes, pigmented pretibial patches (diabetic dermopathy), and bullous eruption of diabetes mellitus (bullosis diabeticorum) – are discussed in detail below.
There are many ways of subclassifying the cutaneous manifestations and associations of diabetes mellitus.410.411.412.413. and 414. The one used here is based on the review published some years ago by Huntley. 415
Vascular and neuropathic complications
Both large and small vessels are affected in diabetes. 415 Atherosclerosis of large vessels contributes to the ischemic complications, such as gangrene of the lower leg, but small vessel (microangiopathic) changes also play an important role. These latter changes are considered below. Other vascular-related phenomena include facial rubeosis411 and the erysipelas-like areas of erythema sometimes seen on the lower parts of the legs, including the feet. Reduced sweating, loss of hair, and glazed skin are, in part, related to vascular changes.
Sensory, motor, and autonomic neuropathies may occur in diabetes. Autonomic dysfunction is sometimes associated with disturbances of sweating and vasomotor phenomena. Neuropathic ulcers may result from the sensory neuropathy. Verrucous skin lesions have developed in association with these neuropathic ulcers on the feet. 416
Infections
Infections are seen less commonly than before, probably because there is better control of diabetes. Bacterial infections that may occur include furuncles, non-clostridial gas gangrene, Pseudomonas infections of the ears, and erythrasma (see p. 554). Infections with Candida albicans are still common in diabetics and may result in paronychia, stomatitis, vulvitis, and balanitis (see p. 588). Dermatophyte infections may not be increased, as previously thought. 417 Rare mycotic infections in diabetics include nocardiosis (see p. 601), cryptococcosis (see p. 590), and the zygomycoses (see p. 603).
Distinct cutaneous manifestations
Diabetes mellitus may be associated with the following conditions: necrobiosis lipoidica (see p. 181), granuloma annulare of the disseminated type (see p. 177), scleredema (also known as diabetic thick skin418– see p. 359), pigmented pretibial patches (see below), bullous eruption of diabetes mellitus (see below), finger ‘pebbles’ which resemble knuckle pads histopathologically419.420.421. and 422. (see p. 816), and eruptive xanthomas (see p. 962). In some diabetics, the skin is waxy and thickened, particularly over the proximal interphalangeal joints of the hands, leading to stiffness of the joints.415. and 423. Another manifestation is yellow skin, due in part to the carotenemia which is present in some diabetics. 411 A reduced threshold to suction-induced blisters has also been found in insulin-dependent diabetics. 424
Less well documented associations415 include skin tags (see p. 814), peripheral edema, yellow nails, and perforating disorders associated with diabetic renal failure and hemodialysis (see p. 324). Patients with diabetes mellitus tend to show a reduced hydration state of the stratum corneum, similar to senile xerosis, but there is no impairment of the barrier function of the stratum corneum. 425
Diabetes mellitus or an abnormal glucose tolerance test is present in a small number of patients with Werner’s syndrome (see p. 328), scleroderma (see p. 304), vitiligo (see p. 283), lichen planus (see p. 38) and Cockayne’s syndrome (see p. 533), and in relatives of patients with lipoid proteinosis (see p. 382).
Diabetes mellitus may occur as a secondary process in the course of a number of diseases, such as hemochromatosis (see p. 390), lipodystrophy (see p. 469), acanthosis nigricans (see p. 504), Cushing’s syndrome, acromegaly, and the hepatic porphyrias, particularly porphyria cutanea tarda (see p. 497). These disorders have their own cutaneous expressions, in addition to any related to the diabetic state.
The use of oral hypoglycemic agents may be complicated by a maculopapular eruption, urticaria, photosensitivity, and flushing when alcohol is consumed (with chlorpropamide) and very rarely by erythema multiforme, exfoliative dermatitis, or a lichenoid eruption.
Localized reactions to the injection of insulin are not uncommon and include allergic reactions, localized induration, anesthetic nodules composed of hypertrophied fat and some fibrous tissue, focal dermal atrophy, ulceration and necrosis, brown hyperkeratotic papules, keloid formation, and localized hyperpigmentation. Insulin-induced lipoatrophy is another complication which may develop 6–24 months after the onset of therapy. 426 It is more common in young females, particularly in areas of substantial fat deposition. The atrophy sometimes occurs at sites remote from injections. 410 Lipohypertrophy presents as a soft swelling resembling a lipoma. It is more common in males. 410 Generalized allergic reactions may also occur; these are more common with beef insulin than pork insulin. 426
Diabetic microangiopathy
Diabetic microangiopathy refers to the abnormal small vessels found in many organs and tissues in diabetes mellitus. The kidneys, eyes, skin, and muscles are particularly affected by this disease process, which is the principal factor determining the prognosis of individuals with diabetes mellitus. 410
Microangiopathy may be involved in the pathogenesis of the pigmented pretibial patches, the erysipelas-like erythema, and the necrobiosis lipoidica that may occur in diabetes mellitus. It may contribute to the neuropathy that sometimes occurs. Small vessel disease may be as important as atherosclerosis of large vessels in producing gangrene of the feet and lower limbs in diabetics. In many instances the microangiopathy is clinically silent.
Histopathology
There is thickening of the walls of small blood vessels in the dermis and subcutis and some proliferation of their endothelial cells. The thickening of the walls and subsequent luminal narrowing is caused by the deposition of PAS-positive material in the basement membrane region. This material is partially diastase labile, although most of it is not. 427 Membranocystic lesions, in which a thin hyaline zone surrounds a small ‘cystic’ space, have been reported in the subcutaneous fat (see p. 471). 428
Granular and homogeneous deposits of C5b-9, along with homogeneous deposits of immunoglobulin within the blood vessels, are a characteristic finding. 429 Similar deposits are found in porphyria cutanea tarda. 429
Electron microscopy
The walls of the small vessels are thickened by multiple layers of veil cells (a fibroblast-like cell) and of basement membrane material.428. and 430. There may also be some deposition of collagen in the walls of small vessels in the dermis. 430
Pigmented pretibial patches
Pigmented pretibial patches (diabetic dermopathy, skin spots) are the most common cutaneous finding in diabetes mellitus, although they are not specific to it.415. and 431. They are found in up to 50% of diabetics, but as they are asymptomatic, they are usually overlooked. 432 The patches are seen more frequently in older patients, and in those who have had diabetes mellitus for a longer period of time. 433 They begin as flat-topped, dull-red papules that are round or oval, discrete or grouped, and situated mainly on the pretibial areas. 410 Involvement of the forearms and thighs has been recorded. 434 As the lesions evolve they develop a thin scale and finally become variably atrophic and hyperpigmented.411. and 426. They vary from 0.5 cm in diameter up to large patches covering much of the pretibial skin.
It has been suggested that the lesions represent an exaggerated response to trauma in skin overlying bony prominences415 and that there may be an underlying diabetic angiopathy.435. and 436. Blood flow levels are considerably higher at the dermopathy sites than at contiguous uninvolved skin sites, refuting the theory that they are ischemic in origin. 437 The presence of pigmented pretibial patches has an unfavorable association with the three most common microangiopathic complications of diabetes mellitus: neuropathy, nephropathy, and retinopathy. 433 A relationship between these patches and coronary artery disease has also been demonstrated. 433
In the early lesions, which are infrequently biopsied, there is edema of the papillary dermis and a mild perivascular lymphocytic infiltrate with some extravasation of red blood cells. 415 There may be mild epidermal spongiosis and focal parakeratosis. Hyaline microangiopathy is invariably present. 436
In atrophic lesions there is neovascularization of the papillary dermis, a sparse perivascular infiltrate of lymphocytes, and small amounts of hemosiderin, mostly in macrophages. Attention has been drawn to the presence of a few perivascular plasma cells in this condition, but plasma cells are almost invariably present whenever there is hemosiderin deposition in the skin. 436 In a recent case studied by the author, hemosiderin was also present in the epidermis, between basal cells and along the basement membrane, a finding not previously recorded.
Bullae, usually multiple and confined to the lower extremities, are a rare complication of long-standing diabetes mellitus. 439 They were originally reported in 1963 as ‘phlyctenar lesions’ because they become dark as they dry up, in the manner of a burn blister. 440 The bullae, which arise on a non-inflamed base, are tense and vary in diameter from 0.5 to 17 cm.426. and 441. They heal in several weeks, usually without scarring.
It seems likely that cases reported under this title do not represent a homogeneous entity, some possibly representing the bullous dermopathy of chronic renal failure.415. and 442. Wang and Ackerman concluded their review on bullosis diabeticorum by supporting this view that it is not a discrete entity. 443 Furthermore, there is an increased frequency of diabetes in patients with bullous pemphigoid. 444
The majority of the bullae reported under this title have been intraepidermal in location, often with spongiotic changes in the surrounding epidermis. 445 It has been suggested that some of these may represent healing subepidermal blisters. In other cases the split has been subepidermal,439.446.447. and 448. immediately beneath the lamina densa. 449 While it has been claimed that these have been early lesions and, as such, may more accurately reflect the true histological picture, 449 it should be noted that in the majority of cases with subepidermal blisters, diabetic nephropathy has been present.442. and 449. The drugs being taken by these patients, a potentially important etiological aspect, have not been listed. In the author’s experience, the bullae have been subepidermal in location.
The bullae contain fibrin and just a few inflammatory cells but there is no acantholysis. There may be diabetic microangiopathy with thickening of the walls of small dermal blood vessels, but there is usually only a sparse perivascular lymphocytic infiltrate. 446
Direct immunofluorescence is negative.
PORPHYRIA
The porphyrias are a clinically and biochemically heterogeneous group of disorders in which there are abnormalities in the biosynthesis of heme, leading to the increased production of various porphyrin precursors.450.451.452.453.454.455.456.457.458.459.460. and 461. The various enzyme deficiencies are not always accompanied by detectable increases in the relevant substrate, and overproduction of the precursors does not necessarily lead to symptomatic disease. However, the homozygous state for a particular enzyme deficiency is probably always clinically overt. The various enzyme deficiencies do not result in heme deficiency because a compensatory increase in substrate concentration is sufficient to restore the rate of heme synthesis to normal. 462
Traditionally, the porphyrias have been classified on the basis of the primary site of overproduction of porphyrins. 463 In the erythropoietic porphyrias (congenital erythropoietic porphyria and erythropoietic protoporphyria) there is disordered synthesis of heme in the bone marrow. 464 In the hepatic porphyrias (acute intermittent porphyria, ALA-dehydratase deficiency, hereditary coproporphyria, variegate porphyria, porphyria cutanea tarda, and hepatoerythropoietic porphyria) there is disordered synthesis of heme in the liver. 463 In some circumstances both sites are involved.
It is more appropriate to classify the porphyrias on the basis of their clinical manifestations. Three categories are then recognized.
1. porphyrias with acute episodes and no cutaneous signs:
acute intermittent porphyria
ALA-dehydratase deficiency
2. porphyrias with acute episodes and cutaneous signs:
hereditary coproporphyria
variegate porphyria
3. porphyrias with cutaneous signs only:
congenital erythropoietic porphyria
erythropoietic protoporphyria
porphyria cutanea tarda
hepatoerythropoietic porphyria.
In addition to these eight types of porphyria, there are rare variants that defy classification. 451 Such cases include neonates who develop a photosensitive eruption due to a transient porphyrinemia resulting from phototherapy for hemolytic disease of the newborn.465. and 466.
The acute episodes referred to above are characterized by abdominal pain, neurological changes, and psychiatric disturbances.451. and 467. Sometimes these acute episodes are precipitated by exogenous factors such as drugs. 468 The cutaneous manifestations are of two types. 469 There may be an acute flare pattern with rapidly evolving, painful and burning lesions with some erythema and edema. 470 This pattern is typical of erythropoietic protoporphyria, although it occurs rarely in some of the other variants (see below). The other cutaneous pattern (seen in hereditary coproporphyria, variegate porphyria, congenital erythropoietic porphyria, porphyria cutanea tarda, and hepatoerythropoietic porphyria) consists of slowly developing skin fragility with the development of blisters, erosions, and scars.470.471. and 472. These aspects are considered in further detail with each particular variant of porphyria (see below).
Biosynthesis of porphyrins
Glycine and succinyl coenzyme A react in the presence of ALA-synthase to form δ-aminolevulinic acid (ALA). 453 This is converted to porphobilinogen and subsequently to protoporphyrin IX by a series of enzymatic reactions (Table 18.1). Protoporphyrin IX is chelated with iron in the presence of ferrochelatase to form heme. 473
Acute intermittent porphyria (OMIM 176000) is an autosomal dominant disorder resulting from a defect in porphobilinogen deaminase (uroporphyrinogen I synthase) which catalyzes the formation of uroporphyrinogen from four molecules of porphobilinogen. 474 The gene encoding this enzyme is at 11q23.3. The disorder is usually latent, but acute episodes consisting of neurological, psychiatric, and abdominal symptoms may be precipitated by various factors, particularly drugs. 475 Barbiturates, sulfonamides, and griseofulvin are most often incriminated. 468 There are no cutaneous manifestations.
Laboratory findings include greatly increased levels of porphobilinogen and ALA in the urine during, and usually between, attacks.
ALA-dehydratase deficiency porphyria
This very rare, autosomal recessive disorder (OMIM 125270) results, as its name indicates, from a deficiency of ALA-dehydratase which was formerly known as porphobilinogen synthase (PBG-synthase). The gene encoding this enzyme is at 9q34. There are intermittent, acute episodes resembling those in acute intermittent porphyria; there are no cutaneous manifestations. 475
The pattern of overproduction of heme precursors closely resembles that seen in severe lead poisoning. There is overproduction of ALA and of coproporphyrinogen III.
Laboratory findings include greatly elevated levels of urinary and fecal coproporphyrins. Porphobilinogen and ALA are increased, as in variegate porphyria (see below), during acute episodes. 476
Variegate porphyria
Variegate porphyria (OMIM 176200), like hereditary coproporphyria, may be associated with acute episodes (as seen in acute intermittent porphyria) and with photocutaneous manifestations (as seen in porphyria cutanea tarda).479. and 480. It is an autosomal dominant disorder in which the activity of the enzyme protoporphyrinogen oxidase (the penultimate enzyme in the pathway of heme biosynthesis) is reduced by approximately 50%. This disorder is quite common in white Afrikaaners and much less so in other white people and in people of other races. 479 The (PPOX) gene, which controls the formation of the responsible enzyme, maps to chromosome 1q22–q23. 481 Homozygous cases of variegate porphyria are rare; they present with severe photosensitivity in childhood. 482 Numerous mutations in this gene have been described in European patients, but in South Africa most patients have inherited the same mutation.483.484.485.486. and 487. A founder mutation has also been reported from Chile. 482 The simultaneous occurrence of symptoms of variegate porphyria has been reported in monozygotic twins. 488
Only a minority of people with the enzyme defect develop clinical manifestations, and only after puberty. The clinical penetrance of variegate porphyria in one large South African family with the R59W mutation was 40%. 489 Acute episodes are precipitated by exogenous factors such as the ingestion of various drugs.468. and 490. Cutaneous changes include skin fragility, blistering and milia formation in sun-exposed areas, as in porphyria cutanea tarda, although very occasionally an acute phototoxic reaction (as seen in erythropoietic protoporphyria) may develop. 470 Variegate porphyria is not usually associated with liver dysfunction, although it has been reported in association with a hepatocellular carcinoma.491. and 492. Dapsone, used in the treatment of dermatitis herpetiformis, has precipitated an acute attack of variegate porphyria. 493
Laboratory findings are somewhat variable, depending on the activity of the disease. 470 There is usually an elevated plasma porphyrin level with plasma fluorescence that is maximal at a wavelength of 626 ± 1 nm. 494 In practice, the diagnosis is most often suggested by finding elevated levels of fecal protoporphyrin and coproporphyrin. 490 Urinary ALA and porphobilinogen are usually increased during acute episodes. Porphobilinogen deaminase activity is sometimes decreased as a secondary phenomenon. 495
It has been reported in one homozygote that the erythrocyte protoporphyrin was raised and that this was predominantly zinc chelated. 496
Congenital erythropoietic porphyria (OMIM 263700) is a rare, autosomal recessive disorder of heme synthesis resulting from a deficiency of uroporphyrinogen III cosynthase (URO-synthase) which leads to an accumulation of porphyrins, particularly uroporphyrinogen I and III, in the bone marrow, blood, and other organs.497.498.499. and 500. The URO-synthase gene has been localized to the narrow chromosomal region 10q25.3–q26.3. 498 Several different mutations have been described; 501 some specific mutations appear to correlate with a milder disease. 502 The defect leads to a chronic photobullous dermatosis, intermittent hemolysis, and massive porphyrinuria. 497 Presentation is usually in infancy with red urine which stains diapers a pink color. 470 Late onset of the disease has been recorded.503. and 504.
The severe photosensitivity leads to the formation of bullae within a day or two of exposure to the sun. Recurrent eruptions may lead to mutilating deformities of the hands and face and sclerodermoid thickening of affected parts. 505 Squamous cell carcinoma of the skin is a rare complication. 506 Sunlight avoidance is the best therapy. 500 Other clinical features include the pathognomonic characteristic of erythrodontia (discoloration of the teeth under normal light and red fluorescence with ultraviolet light), hypertrichosis, patchy scarring alopecia, and nail changes.497.507.508. and 509.
There are large amounts of uroporphyrins in the urine and coproporphyrins in the stool; these are predominantly type I isomers. 505 The plasma and erythrocytes contain increased levels of uroporphyrinogen and, to a lesser extent, coproporphyrinogen. 510 Heterozygotes have blood levels of URO-synthase activity intermediate between affected individuals and controls. 505
In the more severely affected individuals, bone marrow transplantation is potentially curative, but is not without risks.511.512. and 513.
Erythropoietic protoporphyria
Erythropoietic protoporphyria (OMIM 177000), more recently termed simply ‘protoporphyria’,470. and 514. results from a defect in the terminal step of heme synthesis at which protoporphyrin IX and iron combine to form heme.515.516.517.518. and 519. It is associated with a partial deficiency of ferrochelatase (heme synthase) leading to an accumulation of protoporphyrin IX in erythrocytes, plasma, liver, and skin. 520 Although originally regarded as autosomal dominant in inheritance, it is now known that autosomal recessive inheritance occurs in a few cases.518.521. and 522. The gene for ferrochelatase has been cloned and mapped to the long arm of chromosome 18 (18q21.3). Over 110 different mutations have now been described.522.523.524.525. and 526. The presence of liver disease appears to correlate with specific mutations. 525
It generally becomes manifest in early childhood with episodes of acute photosensitivity accompanied by a painful, burning sensation. 516 Often there is edema and erythema and rarely there may be urticaria and petechiae.470. and 527. The changes develop within a few hours of exposure to the sun. In the chronic stages there is waxy scarring of the nose, radial scars around the lips, and pits or scars on the forehead, nose and cheeks.470. and 528. The skin on the dorsum of the hands has a leathery texture and it may also show ‘cobblestone’ thickening. 470 It has a marked impact on quality of life. 520 Deaths from cirrhosis accompanying heavy accumulation of protoporphyrin in the liver have been reported.529.530.531. and 532. Cirrhosis and liver failure occur in less than 5% of patients, but up to 20% may have abnormalities in liver function. 522
Rare clinical presentations have included late onset,528.533. and 534. the presence of scarring bullae and milia, 535 coexistence with lupus erythematosus, 536 a clinical picture resembling hydroa estivale537 (see p. 536), and the presence of a fibrous band involving a digit (pseudoainhum). 538 Late-onset disease often occurs in association with myelodysplastic syndrome, 539 often the sideroblastic anemia subtype. 540 Acquired erythropoietic protoporphyria has been reported in two patients as a result of myelodysplasia causing loss of chromosome 18, the locus of the ferrochelatase gene.540. and 541. In one group of patients with protoporphyria the disease appears to be exacerbated by the ingestion of iron; 515 in rare patients there is symptomatic response to iron supplementation. 542 Exacerbation has also followed blood transfusion. 543 Clinical improvement, with lower erythrocyte porphyrin levels, occurs during pregnancy.544. and 545.
Protoporphyria is the only disorder of porphyrin metabolism with normal urinary porphyrins. 470 There are markedly increased levels of protoporphyrin in the feces and blood. The erythrocytes show a red fluorescence which decays more rapidly than that in congenital erythropoietic porphyria (see above). 470
Advice about sun avoidance and sun-protective measures should always be given. These patients are at risk for the subsequent development of vitamin D deficiency. 546 Narrowband UVB desensitization phototherapy has been given to patients prior to their holidays, so that they might be asymptomatic during such a period. 534 Bone marrow transplantation may also be used for long-term control. 547
Porphyria cutanea tarda
Porphyria cutanea tarda (PCT – OMIM 176100) is the commonest form of porphyria in Europe and North America.470. and 548. Its prevalence ranges from 1:5000 to 1:25000 people. 549 It is not a single disorder but an etiologically diverse group that share in common reduced activity of uroporphyrinogen decarboxylase (UROD), the enzyme which catalyzes the sequential decarboxylation of uroporphyrinogen to coproporphyrinogen in a two-stage process.470.550. and 551. In most forms there is a reduction in hepatic UROD, leading to overproduction of porphyrins in the liver. 552 In the uncommon familial form there is usually a deficiency of this enzyme in erythrocytes as well as in other tissues.
There are three major forms of PCT: familial, sporadic, and toxic. In addition, the porphyria resulting from porphyrin-producing tumors of the liver550. and 553. and that associated with chronic renal failure and UROD deficiency are sometimes categorized as two further clinical variants of PCT.552.554.555.556.557. and 558. Hepatoerythropoietic porphyria (see below) is regarded as the homozygous deficiency of UROD. 552
The familial form (type II) is inherited as an autosomal dominant trait. 552 Numerous different mutations have been described.559. and 560. The enzyme (UROD) is located on chromosome 1p34. 561 The familial form was previously thought to be associated, invariably, with a deficiency of UROD in erythrocytes as well as in the liver, but cases with a normal level of erythrocyte UROD have been documented. 562 Sometimes, overt disease is precipitated by some exogenous factor such as childbirth, exposure to ultraviolet radiation in tanning parlors, iron overload, or excessive alcohol intake.548. and 563. Its onset is usually earlier than that of the sporadic form. 564 Type III PCT is characterized by a family history of the disease, although it is biochemically indistinguishable from the sporadic form. 559
The sporadic form (type I) usually has its onset in mid-life. More than 70% of cases in the past were associated with alcohol abuse and liver damage.552.565. and 566. Now, there is a virtual ‘epidemic’ of cases associated with hepatitis C virus, some of whom also develop hepatocellular carcinoma.567.568.569.570.571.572.573. and 574. The prevalence of hepatitis C infection in one Argentinean study was 35.2%. 549 It has been suggested that the porphyria subtype should be called ‘chronic hepatic porphyria’. 575 Estrogen therapy for carcinoma of the prostate576. and 577. or for menopausal symptoms is sometimes the precipitating event. The menopause itself has also been incriminated; menstruation may act as a natural phlebotomy. 578 Oral contraceptives have also been incriminated. 579 Improvement in the disease has resulted from the use of anastrazole to treat a patient with concurrent carcinoma of the breast. 580 Anastrazole, an aromatase inhibitor, suppresses estrogen. Rare associations have included diabetes mellitus, 565 solar urticaria, 581 Wilson’s disease, 582 hepatocellular carcinoma,550. and 583. lymphoma, 584 HIV infection,585.586.587. and 588. agnogenic myeloid metaplasia, 589 idiopathic myelofibrosis, 590 and lupus erythematosus.591.592. and 593. The sporadic form appears to result from inactivation of UROD in the liver551 although this appears to be independent of, rather than the consequence of, liver injury. 552 Mutations in the HFE gene associated with hereditary hemochromatosis have been associated with sporadic and familial PCT.561. and 594. There is a high prevalence of HFE gene mutations in patients with PCT in the Czech Republic. 595 The C282Y mutation in the HFE gene appears to predispose patients to PCT. 596 Co-inheritance of the genes for these two conditions appears to accelerate the onset of the porphyria.597. and 598. Phlebotomy should be first-line therapy in these patients as HFE C282Y homozygotes do not respond to chloroquine; response is limited to those patients with PCT and HFE wild type. 599
The toxic form results from exposure to polychlorinated aromatic hydrocarbons.561. and 600. Other hepatotoxins have rarely been incriminated.
The cutaneous changes occur predominantly on light-exposed areas such as the face, arms, and dorsum of the hands. 601 Their severity is highly variable. They include increased vulnerability to mechanical trauma and the formation of subepidermal vesicles or bullae measuring 0.5–3 cm in diameter. 565 Erosions, milia, scars, and areas of hyperpigmentation are common. 565 Hypertrichosis, patchy alopecia, darkening of gray hair, 602 infections, 603 dystrophic calcification, and sclerodermoid changes may occur but do so less frequently than the other cutaneous manifestations. 565 The sclerodermoid changes, which may be present in up to 20% of cases, do not always occur in light-exposed areas. They may involve the scalp.604. and 605. They are not associated with sclerodactyly and they are not invariably permanent. It has been suggested that they are related to high levels of uroporphyrinogen. 606
Hepatic changes are common in PCT. There is an increased risk of developing hepatocellular carcinoma. 550
Laboratory findings include increased amounts of uroporphyrins in the urine and plasma and of coproporphyrins in the feces. The urine contains a predominance of 8-carboxyl and 7-carboxyl porphyrins, while the feces contain isocoproporphyrin, which is not found in significant amounts in other porphyrias. 551
Hepatoerythropoietic porphyria
Hepatoerythropoietic porphyria (OMIM 176100, as for PCT) is a rare, severe variant that is manifested clinically by photosensitivity commencing in early childhood.607.608.609. and 610. Fewer than 50 patients have been reported. 611 As in porphyria cutanea tarda (PCT), there is a deficiency of uroporphyrinogen decarboxylase (UROD), but the activity of this enzyme is much less than in PCT, reflecting a homozygous state.612. and 613. Several different point mutations and/or a deletion in the UROD gene on chromosome 1p34 have been reported. 614 Hepatic involvement is common. The cutaneous features resemble those of PCT. 615
Laboratory findings are similar to those in PCT, but an additional feature is the presence of elevated levels of protoporphyrins in erythrocytes. 607
Pseudoporphyria
The term ‘pseudoporphyria’ is used for a phototoxic bullous dermatosis which resembles porphyria cutanea tarda (PCT).616. and 617. However, there are normal levels of porphyrins in the serum, urine, and feces. 618 The term ‘therapy-induced bullous photosensitivity’ has been suggested as more appropriate619 as this condition has been reported following the use of a number of drugs including tetracyclines,620. and 621. β-lactams (ampicillin-sulbactam and cefepime) 622 sulfonamides, isotretinoin, 623 ketoprofen, 624 pravastatin, 625 bumetanide, furosemide (frusemide),626. and 627. torsemide, 628 nalidixic acid, 629 ciprofloxacin, 630 voriconazole,624. and 631. naproxen,632.633.634. and 635. celecoxib, 624 rofecoxib, 636 amiodarone, 631 oxaprozin, 637 mefenamic acid, 638 aspirin, 631 nabumetone,639.640.641.642. and 643. pyridoxine, 644 chlorthalidone, 645 the oral contraceptive pill, 646 flutamide647 cyclosporine (ciclosporin), 5-fluorouracil, excessive cola consumption, 631 acitretin, 648 and etretinate. 649 A few cases have followed the prolonged use of tanning beds.650.651. and 652. Blistering has also been reported in patients with chronic renal failure undergoing hemodialysis.653. and 654. In one case it followed the use of narrowband UVB phototherapy, used in the treatment of psoriasis. 655 In some patients pseudoporphyria has been associated with a deficiency of uroporphyrinogen decarboxylase with increased levels of porphyrins; in others, many of whom were also receiving furosemide (frusemide), the porphyrin levels have been normal.653.656. and 657. The various causes are listed in Table 18.2.
Only gold members can continue reading. Log In or Register to continue