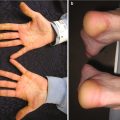
Fig. 1.1
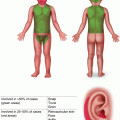
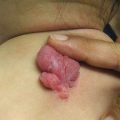
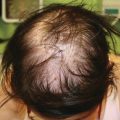
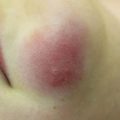
Characteristic appearance of a Spitz nevus as a smooth dome-shaped pink papule [16]. Reprinted from Journal of the American Academy of Dermatology, Vol. 65, No. 6, Luo S, Sepehr A, Tsao H, Spitz nevi and other Spitzoid lesions: Part I. Background and Diagnoses, pages 1073–1084, © 2011, with permission from Elsevier
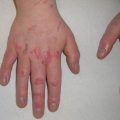
On dermoscopy, Spitz nevi have several common patterns, including starburst, globular, homogeneous, negative network, and reticular patterns (Fig. 1.2) [17]. The starburst pattern is demonstrated in more than 50% of biopsied Spitz nevi and is associated with 96% diagnostic sensitivity. This pattern was named for its exploding star-type appearance and can be composed of streaks (pseudopods or radial streaming) or multiple rows of peripheral globules [18]. The starburst pattern signifies an initial radial growth phase before transitioning to a homogeneous pattern with a blue-white veil [19]. The starburst pattern also characterizes pigmented spindle cell nevi [20]. Common melanoma-specific structures, such as atypical pigment network, negative network, crystalline structures, blue-white veil, and irregular vessels, have been described in Spitz nevi but are more likely to be symmetric and organized.

Fig. 1.2
Common dermoscopy patterns seen in Spitz nevi [17]. Reprinted from Dermatologic Clinics, Vol. 31, No. 2, Kerner M, Jaimes N, Scope A, Marghoob AA, Spitz nevi: a bridge between dermoscopic morphology and histopathology, pages 327–335, © 2013, with permission from Elsevier
Histology
While the majority of Spitz nevi are compound, they can also be junctional or intradermal. At low power, Spitz nevi are dome or wedge shaped, symmetric, and well-circumscribed. At higher power, Spitz nevi have characteristic epithelioid and/or spindled melanocytes with prominent cytoplasm. Junctional cells are nested and often have clefts separating individual cells or nests from adjacent epidermis. Cells mature to become small individual melanocytes deeper in the dermis. Kamino bodies are acellular pale eosinophilic structures often found within the epidermis and are composed of basement membrane material. Epidermal hyperplasia, hyperkeratosis, and hypergranulosis are commonly seen. Occasional mitotic figures can be seen within the epidermis or superficial dermis, although deep mitoses can indicate a more atypical tumor (Fig. 1.3a, b ). Pigmented spindle cell nevi resemble Spitz nevi histologically but have prominent cytoplasmic pigmentation.
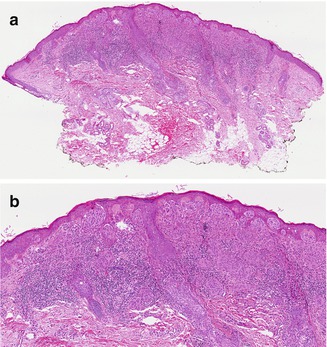
Fig. 1.3
Spitz nevus. (a) At low power, the characteristic wedge-shaped, symmetric, well-circumscribed architecture of a Spitz nevus is seen (H&E, 40×). (b) Junctional and dermal nests of mostly epithelioid and some spindled melanocytes are seen. Eosinophilic globules ( Kamino bodies) are in the epidermis, and there is mild epidermal hyperplasia. Lymphohistiocytic inflammation is present in the dermis (H&E, 100×) (Image courtesy of Adam I. Rubin, MD)
Genetics
Approximately 20% of Spitz nevi demonstrate an increase in chromosome 11p, which is the site of the HRAS gene [21]. Gain of this genetic locus is very rare in cutaneous melanoma. Gain of chromosome 11p therefore suggests a more benign etiology (see Genetics and Molecular Analysis below). Apart from an occasional gain in HRAS, genetic gains or losses are rare in Spitz nevi.
Management
Although generally considered to be benign, controversy exists surrounding the management of Spitz nevi. For classic appearing Spitz nevi in school-aged children, some advocate for clinical observation, while others advocate a biopsy of any Spitzoid lesion [22–29]. There is consensus throughout the literature, however, that biopsy should be performed for any lesion with atypical features, rapid evolution, asymmetry, or ulceration. A partial biopsy should be avoided because overall architecture is an important criterion for diagnosis of a Spitz nevus. If clinically visible tumor remains after initial biopsy, then most clinicians would recommend re-excision. If only the histologic margins of a biopsy are positive, the need for re-excision is controversial. In a survey of pediatric dermatologists, approximately one-third of respondents would re-excise a Spitz nevus after partial biopsy, which is similar to the results of a survey of general dermatologists [28–30]. Though, if re-excision is pursued, narrow 1–2 mm margins are adequate.
Atypical Spitz Tumors
Key Points
Histologic features overlapping with Spitz nevi and melanoma.
Molecular testing with fluorescence in situ hybridization and/or comparative genomic hybridization is an adjunct to histologic evaluation.
HRAS mutations, BAP1 mutations , and kinase fusions have been identified in these tumors.
Atypical Spitz tumors (AST), also called Spitzoid tumors of uncertain malignant potential (STUMP) , are challenging because they have histologic features that overlap with both classic Spitz nevi and with melanoma. Consequently, the diagnosis and management of these tumors are controversial and still evolving.
Atypical Spitz tumors are often considered within the larger category of “borderline melanocytic tumors ” or “melanocytic tumors of unknown malignant potential” (MelTUMP). The term “MelTUMP ” was first used by Elder and Xu to encompass a wide spectrum of lesions that demonstrate features of both benign melanocytic nevi and melanoma [31]. In addition to AST, pigmented epithelioid melanocytoma, deep penetrating nevus, and cellular blue nevus are the most common neoplasms included in this group.
Clinical
Atypical Spitz tumors have a varied clinical appearance. They can be indistinguishable from benign Spitz nevi and present as growing pink, red, brown, or black papules. In other cases, AST can have concerning clinical characteristics, such as rapid growth, irregular pigmentation, ulceration, nodularity, or easy bleeding. AST can be found anywhere on the body but are most often on the trunk or extremities [32].
Genetics
Several mutations have been identified in ASTs, including HRAS mutations, BAP1 mutations, and kinase fusions. These mutations are not unique to AST and are often not definitive for distinguishing an AST from a Spitz nevus or melanoma. Comparative genomic hybridization (CGH) and fluorescence in situ hybridization (FISH ) are being used to further identify chromosomal aberrations in tumors to aid in diagnosis (see Molecular Analysis below).
ASTs with HRAS mutations have an increase in chromosome 11p copy number and consequently HRAS gain of function. HRAS is an oncogene that activates both the MAP/ERK and PI3K/AKT/mTOR pathways allowing for cell proliferation. HRAS mutations are rarely found in melanoma [33–36] and, when found in AST, tend to have a good prognosis [37]. On histology, these tumors are predominantly intradermal and comprised of large, pleomorphic melanocytes that have an infiltrating growth pattern at the base. There is marked stromal desmoplasia and sclerosis [35].
BAP1 (BRCA1-associated protein 1) is a nuclear protein that functions as a tumor suppressor and has a role in DNA damage repair, cellular differentiation, transcription, and cell cycle control. Germline BAP1 mutations are found in cancer predisposition syndrome, but somatic loss of BAP1 has been identified in some ASTs. These tumors generally also harbor BRAF mutations [38]. BAP1 inactivated tumors have also been known as BAP1-inactivated Spitzoid nevus, BAP-oma, nevoid melanoma-like melanocytic proliferation (NEMMP) , and melanocytic BAP1-mutated atypical intradermal tumors (MBAIT) [39]. BAP1 inactivated tumors tend to be skin-colored or dome-shaped papules or nodules. Histologically, BAP1-inactivated tumors are predominantly dermal tumors comprised of large epithelioid cells with abundant amphophilic cytoplasm, nuclear pleomorphism, and prominent nucleoli [39]. Loss of BAP1 staining on immunohistochemistry can be used to identify these tumors.
Kinase fusion proteins involving the tyrosine kinases ALK, ROS1, NTRK1, RET, and MET and the threonine kinase BRAF have been identified in approximately 50% of AST [40, 41]. The fusion of these kinase genes with another gene creates an enzyme that is constitutively active. ALK fusion proteins are present in 5–15% of AST and are most commonly fused with TPM3 and DCTN1 [41–43]. ALK-positive tumors typically have a plexiform growth pattern with fascicles of fusiform melanocytes in the dermis [42, 43]. ALK immunohistochemical staining is positive in these tumors. ROS1 fusion proteins are present in 6% of AST [41]. These tumors have no characteristic clinical or histologic appearance, and ROS1 staining has low sensitivity and specificity. NTRK1 fusion proteins have been identified in up to 25% of AST [40]. NTRK1 staining has high sensitivity and specificity and can be useful in identifying ASTs that otherwise have no characteristic clinical or histologic findings. RET, MET, and BRAF fusions are less commonly found in AST [40, 41, 44].
Histology
Histopathologic criteria for AST have not been established. There is frequently variability between pathologists as to the whether a tumor is a benign Spitz nevus or an AST and whether a tumor is an AST or Spitzoid melanoma. In one study, there was poor agreement between 13 expert dermatopathologists asked to evaluate a cohort of atypical tumors [45]. Molecular genetic analyses have been pursued as a way to more precisely define and distinguish between these entities.
Compared to the classic histology of Spitz nevi, AST may demonstrate greater cytologic atypia, ulceration, cellular density, lack of maturation, deep mitoses, and larger size. Histologic features that would be considered more consistent with a diagnosis of melanoma include a mitotic rate above 6 mitoses/mm2, significant asymmetry, and tumor extension to the subcutaneous fat [46].
Immunohistochemical stains for HMB-45 (Gp-100), Ki-67 (MIB-1), and p16 can be used together to further assess these tumors. HMB-45 is a melanocyte stain that can be used to assess the maturation of a tumor. Spitz nevi demonstrate maturation with depth, so HMB-45 expression decreases toward the base. On the other hand, melanoma demonstrates a more uniform or scattered HMB-45 distribution throughout. Ki-67 is a marker of cellular proliferation and in benign nevi is usually present in the epidermis but is absent in the deep dermis, also reflecting melanocyte maturation. Increased Ki-67 expression in deep dermal melanocytes is seen in melanoma [47]. CDKN2A on chromosome 9p21 encodes for the tumor suppressor p16 and has been found to be associated with more clinically aggressive AST [44, 48–51]. If staining for p16 is lost, it is indicative of a homozygous loss of 9p21 and may be associated with more aggressive behavior [44].
Molecular Analysis
As the genetic landscape of melanocytic tumors has become more defined, chromosomal analysis is used to distinguish between Spitz nevi, AST, and melanoma. FISH and array CGH are molecular techniques that identify genetic aberrations in tumors. FISH utilizes fluorescently labeled short fragments of DNA (probes) that bind to tumor DNA. Each nucleus should bind two probes and thus have two fluorescent dots. If there is chromosomal gain or loss corresponding to the probes, then there will be more or less than two dots, respectively. Array CGH is similar, though able to detect chromosomal gain and loss over the entire genome through binding to a DNA microarray.
Current FISH testing includes the five probes, RREB1/6p25, MYC/8q24, CDKN2A/p16/9p21, CCND1/11q13, and Cen9/centromere 9. This set of probes have been shown to have a sensitivity of 94% and specificity of 98% for detecting Spitzoid melanomas [52]. Studies have found clinical patterns based on FISH testing that can help prognosticate AST (Table 1.1).
Table 1.1
Risk stratification of atypical Spitz tumors based on fluorescence in situ hybridization results
High risk | Intermediate risk | Low risk |
|---|---|---|
Isolated homozygous loss of 9p21 | Isolated gain 6p25 or gain 11q13 | Heterozygous loss 9p21 Isolated loss of 6q23 Negative FISH |
AST with isolated homozygous loss of 9p21 exhibit more aggressive behavior, with a greater likelihood of tumor spread to lymph nodes and death due to disease [44, 48–51]. Because of this aggressive behavior, the term “Spitzoid melanoma with isolated homozygous loss of 9p21” has been proposed for these tumors [44, 48]. Although more aggressive, these tumors seem to have a better prognosis than conventional melanomas [44, 48]. AST with isolated heterozygous loss of 9p21 have a more benign clinical course, with no tendency for metastasis and with a prognosis similar to tumors with negative FISH [44, 48–51]. Tumors with isolated heterozygous loss of 9p21 have less cytologic atypia, less atypical dermal mitoses, less nodular growth, and more Kamino bodies compared to those with homozygous loss of 9p21 [44]. Gain of 6p25 or 11q13 also exhibits aggressive behavior but may be less aggressive than tumors with homozygous 9p21 loss [48]. Gain of 8q24 has been rarely identified in AST, appears as amelanotic papules or nodules, and histologically has sheets of small- to intermediate-sized melanocytes with monotonous cytologic appearance, nuclear atypia, and prominent mitoses [49]. Because these tumors are so rare, it is difficult to assess their risk. In the past, a probe for MYB/6q23 was included in FISH panels but has been replaced with 8q24. Though AST with isolated loss of 6q23 could have positive sentinel lymph nodes, these tumors tend not to spread beyond the sentinel node or have metastasis [48, 49, 53].
Array CGH identifies copy number alterations over the entire genome of a tumor. In general, benign nevi lack copy number alterations, while melanomas have multiple copy number alterations, sometimes involving a portion of a chromosome but other times entire chromosomes [54, 55]. Melanomas often have amplification of oncogenic genes (BRAF/7q34 and MITF/3p13) or gain in larger oncogenic regions (6p, 7, and 8q). Homozygous loss of tumor suppressor genes (CDKN2A/9p21 and PTEN/10q23) and tumor suppressor regions (6q, 8p, 9p, 10) are commonly seen. The more atypical Spitzoid tumors have more chromosomal aberrations.
Management
No guidelines for management exist for these extremely challenging tumors. Because AST have an uncertain malignant potential, complete excision is recommended. The role of sentinel lymph node (SLN) biopsy for the diagnosis and management of AST is controversial. Some have advocated for the use of SLN biopsy to aid in diagnosis, with the presence of tumoral deposits in the lymph node as evidence for malignancy. The interpretation of finding atypical cells in the SLN is complicated by the fact that deposits of benign nevi, including Spitz and blue nevi, can be found within local lymph nodes [56–60]. Furthermore, there is mounting evidence that SLN biopsy is not useful for diagnosis or management for AST. Even when the SLN is positive, multiple studies have found that it does not indicate an increased risk for metastasis or increased mortality [61–66]. In pediatric patients with AST treated with excision with clear margins alone and without SLN biopsy, a follow-up study showed excellent prognosis, with no recurrence, metastasis, or death from disease [65]. Patients with a history of AST should continue to have close clinical follow-up 1–2 times a year.
Pediatric Melanoma
Key Points
Uncommon and decreasing in incidence.
Three age categories: congenital (in utero to birth), childhood (<10 years of age), and adolescent (10–19 years of age).
Additional ABCD criteria of pediatric melanoma (amelanotic, bleeding, bump, color uniformity, de novo, of any diameter) aids in identifying suspicious lesions.
Pediatric melanoma is an uncommon diagnosis and accounts for about 6% of cancers in adolescents between 15 and 19 years of age. The incidence of melanoma in individuals under 20 years of age is 0.37 per 100,000 person years, with the incidence increasing with age [67]. Fortunately, the incidence of pediatric melanoma has been decreasing up to 11% annually in the United States [68–70]. This decrease may be due to improved public health programs educating about the risks of indoor tanning and advocating for sun safety and sun protection. After Australia instituted a skin cancer educational program, a similar decrease in incidence was documented [71].
Clinical
Pediatric melanoma can be subdivided into three age groups: congenital (in utero to birth), childhood (<10 years of age), and adolescent (10–19 years of age). Congenital melanoma is extremely rare, with only 23 cases reported between 1925 and 2002 [72]. It can occur within a congenital melanocytic nevus or secondary to transplacental transmission of maternal melanoma. Childhood and adolescent melanoma can occur de novo or in association with another nevus.
The clinical presentation of melanoma in children differs from that in adults. A study evaluating the usefulness of the classic ABCDE criteria (asymmetry, irregular borders, multiple or irregular color, diameter >6 mm, evolution) in the pediatric population found it failed to identify melanoma in 60% of cases in patients less than 10 years of age. Additional ABCD criteria were recommended in this prepubertal age group: amelanotic, bleeding, bump, color uniformity, de novo, of any diameter [73].
Overall, there is a female preponderance for pediatric melanoma, though more common in males in the prepubertal (<10 years of age) age group and females in adolescence [74]. Tumors in the younger age group tend to be significantly thicker and diagnosed at a more advanced stage than in adolescence [74]. This difference is likely related to the delay in diagnosis because of the low index of suspicion for melanoma in younger age groups and difficulty or reluctance to biopsy children. Head and neck melanoma is more common in the very young, while the trunk is more common in adolescence [69].
Risk Factors
Risk factors for the development of melanoma in children are fair skin, blue or green eyes, blond or red hair, tendency to freckle, intermittent intense sun exposure, family history of melanoma, personal history of atypical nevi, and systemic immunosuppression. Additional risk factors include large congenital melanocytic nevi, familial atypical multiple mole melanoma syndrome, BAP1 tumor predisposition syndrome, and xeroderma pigmentosum.
Congenital melanocytic nevi (CMN) are categorized based on their projected final adult size, with small CMN measuring less than 1.5 cm, medium-sized CMN 1.5–20 cm, and large CMN greater than 20 cm. Small and medium CMN have a lifetime risk of less than 1%, though the general risk of melanoma in the United States is about 2% [67, 75–81]. The reported risk of melanoma in large CMN ranges from 0 to 50%, though the risk is likely closer to 5–10% [77, 82–92]. Importantly, prophylactic excision of CMN does not entirely eliminate the risk of melanoma [91, 93].
Familial atypical multiple mole melanoma (FAMMM) syndrome is characterized by the development of atypical nevi starting early in life and an increased lifetime risk of melanoma. Inherited, in an autosomal dominant manner, FAMMM is caused by a mutation in CDKN2A, which encodes for p16, a tumor suppressor. In addition to the risk for cutaneous melanoma, patients with FAMM have an increased risk of developing ocular melanoma and other internal malignancies, most commonly pancreatic cancer. Up to 10% of melanomas in this group occur in individuals before the age of 20, making the identification of at-risk children imperative [94]. Melanoma astrocytoma syndrome (MAS) is a variant of FAMMM that is also caused by a mutation in CDKN2A. CDKN2A encodes for two proteins, p16 and p14, both tumor suppressors, and MAS is caused by a mutation in p14. Patients with MAS are at risk for melanoma and for a number of central nervous system tumors, including astrocytoma and meningioma.
As noted above, mutations in BRCA1-associated protein 1 (BAP1) has been identified as a risk factor for multiple malignancies. In addition to characteristic BAP1-inactivated tumors described above (see Atypical Spitz Nevus), patients with BAP1 tumor predisposition syndrome develop cutaneous and uveal melanomas, mesothelioma, clear cell renal cell carcinoma, and basal cell carcinoma.
Xeroderma pigmentosum (XP) is a rare autosomal recessive disorder caused by mutations in the DNA repair mechanism, so patients are unable to repair UV-induced DNA damage. Patients with XP are at an increased risk of developing multiple types of skin cancer, including melanoma. Melanoma develops in approximately 5% of patients with XP, often at younger than 20 years of age [95].
Genetics
Pediatric conventional melanoma has the greatest number of single-nucleotide variations of any pediatric cancer [96]. Mutations in TERT (telomerase reverse transcriptase core promoter) are found in all pediatric conventional melanoma [96]. Eighty-seven percent of conventional melanoma also has BRAF V600 mutations , commonly in association with a PTEN mutation [96]. Interestingly, BRAF V600E mutations have been found in a higher proportion of adolescents with histologically conventional melanomas than in the adult population [97]. Melanoma arising in a congenital nevus tends to have mutations in NRAS. Some Spitzoid melanomas have kinase fusions, and another subset with TERT mutations is at increased risk for metastasis and poor prognosis [96, 98].
Management
Staging of pediatric melanoma utilizes the adult staging system recommended by the American Joint Commission on Cancer (Tables 1.2 and 1.3) [99]. All melanoma should be completely excised, with excision margins identical to those recommended for melanoma excision in adults (0.5 cm for in situ, 1 cm for tumors <1 mm thick, 1–2 cm for tumors 1–2 mm thick, and 2 cm for tumors >2 cm thick) [100, 101].
a. Tumor classification | ||
Classification | Thickness (mm) | Ulceration status/mitoses |
Tis | N/A | N/A |
T1 | ≤1 | a: Without ulceration and mitoses <1mm2 b: With ulceration or mitoses ≥1mm2 |
T2 | 1.01–2 | a: Without ulceration b: With ulceration |
T3 | 2.01–4 | a: Without ulceration b: With ulceration |
T4 | >4 | a: Without ulceration b: With ulceration |
b. Regional lymph node classification | ||
Classification | Number of metastatic nodes | Nodal metastatic burden |
N0 | None | N/A |
N1 | 1 | a: Micrometastasis b: Macrometastasis |
N2 | 2–3 | a: Micrometastasis b: Macrometastasis c: In-transit met(s)/satellite(s) without metastatic nodes |
N3 | 4+ Metastatic nodes, matted nodes, in-transit met(s)/satellite(s) with metastatic node(s) | |
c. Distant metastasis classification | ||
Classification | Site | Serum LDH |
M0 | No distant metastases | N/A |
M1a | Distant skin, subcutaneous, or nodal metastases | Normal |
M1b | Lung metastases | Normal |
M1c
Related posts:Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

| ||