Mechanisms of Wound Repair, Wound Healing, and Wound Dressing: Introduction
|
Introduction
The terms “wound healing”, “wound repair”, or “tissue repair” are often used interchangeably, but actually “healing” and “repair” point to different sets of events and outcomes. First of all, before any distinction is made, one must recognize the rather obvious fact that “healing” and “repair” are not confined to the skin, but can encompass any organ system. Technically, wound healing is a term that should be used only in the context of true regeneration, when the original architecture and structure of an organ or anatomic part is completely restored to the way it was before injury. More primitive animals, such as small amphibians and reptiles, are still capable of this type of regeneration. However, as animals became larger and more complex during evolution, true regeneration was no longer possible. The human fetus is still largely capable of regeneration (especially in the early stages) but, in adults and with the possible exception of the liver (probably a compensatory enlargement and not regeneration), true regeneration does not take place. Rather, man and other higher vertebrates heal by a process of repair (wound repair or tissue repair), whereby the eventual outcome is not true anatomic restoration but a functional compromise. Still, because of well established terms and the published literature, even within our discussion here, we may at times use “healing” and “repair” interchangeably; we will be more specific when truly referring to the process of tissue regeneration.
There are probably evolutionary reasons for why repair occurs in higher vertebrates compared to true healing or regeneration. Teleologically and from an evolutionary standpoint, the process of repair for higher animals needed to be rapid, economical from an energy standpoint, and allow for the immediate survival of the organism. However, by necessity, repair leads to a rapid solution to injury and thus to scarring. Another important consideration is that most of the mechanisms of wound repair that have evolved are aimed at addressing acute tissue injury and not chronic conditions. Again from an evolutionary standpoint, humans were not meant to develop degenerative diseases or live long enough to develop arterial, venous and pressure ulcers, or neuropathic ulcers from diabetes. Therefore, humans are quite unprepared for these types of chronic wounds, and there are no specific mechanisms that have evolved to deal with them in a truly effective way.1–3 It should be noted that the extent of injury and its depth are important considerations. Thus, shallow wounds (i.e., shave biopsies) that retain skin appendages (hair follicles, sweat glands, etc.) in the wound bed have the capacity to heal from the “inside”, since keratinocytes associated with those skin appendages are still present. On the other hand, full-thickness wounds (punch biopsies are a good example) have to rely on keratinocyte migration and proliferation from the edges (Fig. 248-1). Not surprisingly, full-thickness wounds are associated with delayed healing and more scarring.2 Some animal models, particularly those in which certain genes are overexpressed or knocked-out, are providing greater understanding of the role of certain proteins and mediators.4–7 Impaired healing is found in mice with combined deficiency of molecules critical in inflammation (E- and P-selectins), and in mice without plasminogen, uPA and tPA (double knock-out), fibroblasts growth factor-2 (basic FGF or bFGF), or inducible nitric oxide (iNOS). On the other hand, impaired or delayed healing occurs in transgenic mice overexpressing some tissue metalloproteinases (MMP-1) and antisense to CD44, the receptor for hyaluronic acid. Some induced mutations lead to accelerated healing, as reported with Smad-3 or skn-1a knock-out mice.8 In the future, it may be possible to use these clues to stimulate the healing process in humans.
Figure 248-1
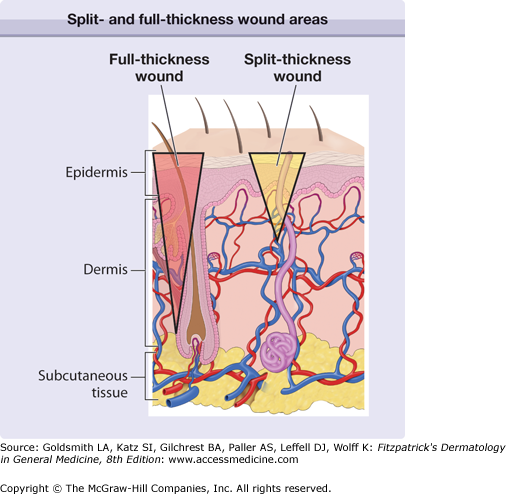
Diagrammatic representation of the skin, with two inverted triangles representing either a split- or full-thickness wound. Extending the injury below the reservoir of keratinocytes present in skin appendages (full-thickness wound) removes the ability for the keratinocytes to populate the defect from within the wound bed; healing has to occur from the wound edges and, moreover, more scarring takes place.
Phases of Wound Healing
In general, there are four recognized phases that characterize the cutaneous repair process: (1) coagulation, (2) inflammatory phase, (3) proliferative and migratory phase (tissue formation), and (4) remodeling phase. The coagulation and inflammatory phases ore sometimes grouped together, so great is the overlap of mediators that are released. In a diagrammatic form, Fig. 248-2 illustrates these phases of healing in sequence, while Fig. 248-3 shows specific events that take place during the different phases. We will discuss these phases and identify the main components and events that characterize them. The cell types primarily involved in wound healing have been regarded to be the platelets, neutrophils and macrophages, fibroblasts, endothelial cells, epithelial cells. More recently, increasing importance is accumulating for the role of lymphocytes, either directly or indirectly.9 However, it should be noted that breaking down the overall process of wound repair into these seemingly defined phases is artificial, as they overlap considerably.2,3 Another consideration is that the occurrence and hypothesis of these phases of wound healing has been determined in laboratory animal experiments; it is assumed that the same processes are involved in human tissue repair.
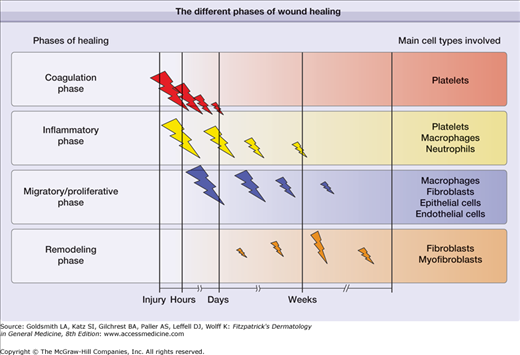

Although we have shown these as separate phases, the degree of overlap is such that it is appropriate to discuss them together. These early phases begin immediately after an acute injury. Disruption of blood vessels leads to local release of blood cells and blood borne elements resulting in clot formation. While the blood clot within the vessel lumen provides hemostasis, the clot within the injury site acts as a provisional matrix for cell migration,10 further formation of new extracellular matrix (ECM),11 and a reservoir for cytokines and growth factors.12 The initial component of this phase is dominated by the platelet, which directs clotting of the fresh wound by the intrinsic and extrinsic pathways. Platelets also release a number of chemotactic factors that attract other platelets, leukocytes, and fibroblasts to the site of injury. Leukocytes are slowed down within the blood stream through the expression of selectins, which, coupled with integrins, bring inflammatory white cells into the wound.5 These cells have multiple roles, including debridement of necrotic material and bacteria, as well as the production of certain critical cytokines. A case in point is the production of connective tissue growth factor (CTGF) by inflammatory cells and expressed in wounds.13 After the first few days, the neutrophils are removed by macrophages.
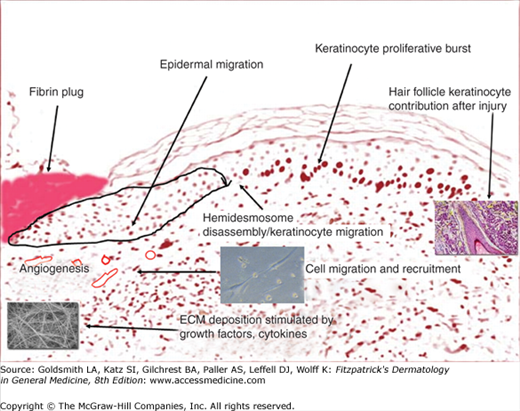
Fig. 248-4 is a diagrammatic representation, using a tissue section, of the events that take place after injury. The fibrin plug resulting from the initial injury provides a temporary wound coverage and consists of platelets embedded within a complex meshwork of mainly polymerized fibrinogen (fibrin), fibronectin, vitronectin, and thrombospondin.3,14 Cross-linking of fibrin by Factor XIII also appears to be critical, and there is evidence that the function and probably migration of keratinocytes is impaired by uncross-linked fibrin.15 As they immediately aggregate, platelets release a wide range of growth factors, including platelet-derived growth factor (PDGF) and transforming growth factor-β1 (TGF-β1). In fact, platelets are the main storage site for the TGF-β1 isoform. The acute injury environment, with its hypoxia, proteases, and low pH, contributes to the activation of these growth factors.2 The principal mediators themselves produce others from their fragmentation or polymerization. A classic example is the chemotactic fibrinopeptides A and B, which are produced from the action of thrombin on fibrinogen.15,16 Another example is the formation of bradykinins, as well as C3a and C5a, which are activated by Hageman factor.17
As the inflammatory component of this early phase continues, within 24–48 hours after injury, monocytes replace neutrophils and become the predominant leukocyte. Monocytes are attracted to the injury site by some of the same chemoattractants responsible for recruitment of neutrophils, such as kallikren, fibrinopeptides, and fibrin degradation products.18 Other, more specific chemoattractants then take over in recruiting monocytes, and they include fragments of collagen, fibronectin, elastin, and TGF-b1. Monocytes undergo a phenotypic change to tissue macrophages and, unlike neutrophils, they are critical for the progression of wound healing.19,20 Macrophages phagocytose and kill bacteria, and scavenge tissue debris.21 They also release several growth factors, including PDGF, fibroblast growth factor (FGF), TGF-b, thereby stimulating migration and proliferation of fibroblasts, as well as production and modulation of extracellular matrix. The macrophage is generally regarded as the master cell in wound healing.22 However, this is an oversimplification, as it is important to consider that what may delay or impair healing is not so much the presence or absence of inflammation and certain cells but, rather, an inappropriate inflammatory response.17 For example, there is evidence that healing may occur in the absence of an inflammatory infiltrate.23,24 Conversely, experiments in mice constitutively expressing the chemotactic cytokine IP-10 show that an intense inflammatory infiltrate can impair neovascularization and the formation of appropriate granulation tissue.25 Therefore, the true role of inflammation in tissue repair remains somewhat controversial from an experimental point of view.26,27 From the clinical standpoint, one observes that in certain cutaneous wounds, such as pemphigus or pyoderma gangrenosum, downregulation of inflammation with the use of corticosteroids is effective. Possibly, modulation and “correction” of the inflammatory response by corticosteroids may be helpful in these selected clinical entities.28
Fig. 248-4 illustrates a representation of the fibrin plug in place and certain events that occur in the next two phases of healing, the proliferative/migratory and remodeling phases. As inflammation has already played its major role early on after injury, the most important event in cutaneous wound healing now becomes reepithelialization. This critical event is not based on keratinocytes alone, because there is a high interdependence between keratinocyte movement through the provisional fibrin matrix, recruitment of fibroblasts and endothelial cells, and extracellular matrix formation (Figs. 248-2 and 248-3). Tissue matrix metalloproteinases (MMPs) and other enzymes (tPA and uPA) are critical to the movement of cells through provisional structural matrix components, as well as for freeing the keratinocytes at the edge of the wound from their hemidesmosomal and desmosomal attachments. Another critical set of molecules that help guide this migratory components are the integrins.29,30 The integrins, consisting of at least 24 αβ heterodimers (18 α and 8 β subunits), are transmembrane cell surface receptors that bind the extracellular matrix (ECM) to cytoskeletal structures.31 The integrin profile is very dynamic during the repair process. For example, dermal fibroblasts undergo a switch from α2 to α3 and α5 integrin subunits. As another example, endothelial cells cannot respond to angiogenic stimuli without the expression of αvβ5 integrin. Certain polypeptide growth factors are absolutely essential to angiogenesis, including basic fibroblast growth factor (bFGF) and vascular endothelial growth factor (VEGF).3,31 Table 248-1 shows some of the main cytokines and growth factors shown to play a role in the repair process. One must realize that such tables are oversimplifications of very complex biologic properties of these polypeptides. Only the main effects are noted, and it must be borne in mind that the actions of growth factors are often context specific and not the same in every biological situation.
Responses | EGF | FGF | GMCSF | IL-1 | PDGF | TGF-β1 | VEGF | CTGF | IGF |
|---|---|---|---|---|---|---|---|---|---|
Fibroblast proliferation | + | + | + | ++ | + | + | + | ||
Keratinocyte proliferation | + | + | + | – | + | ||||
Angiogenesis | + | + | + | + | + | + | ++ | ||
Matrix formation | + | ++ | ++ | ++ | + | ||||
Inflammatory cell migration/chemotaxis | ++ | + | + |
Evidence points to low oxygen tension as an important early stimulus for fibroblast (and endothelial cell) activation. Fibroblast replication and longevity are enhanced in hypoxia,32 and low oxygen tension stimulates clonal expansion of dermal fibroblasts seeded as single cells.33 Moreover, the synthesis of a number of growth factors is enhanced in hypoxic cells. Macrophages secrete an angiogenic substance only when they are exposed to low oxygen tension. This reversible effect was observed at an oxygen tension of 15–20 mm Hg.34 TGF-β1 transcription and peptide synthesis are enhanced in cultures of human dermal fibroblasts exposed to a similar level of hypoxia.35 In addition, hypoxia upregulates the synthesis of endothelin-1,36 PDGF B chain,37 and VEGF38 in endothelial cells. It appears that, at least in some cases, the effect of hypoxic conditions is mediated by hypoxic inducible factor-1 (HIF-1), a DNA binding complex shown to contain at least two basic helix-loop-helix PAS-domain proteins.39
The importance of the hemidesmosome and desmosome is well known for many dermatological diseases where specific defects in their overall structural integrity are present (i.e., pemphigus, bullous pemphigoid, epidermolysis bullosa, etc.). Now, however, during the healing process and for keratinocyte migration to occur, there is a need to break down these complex structures anchoring the basal keratinocytes to the basement membrane and neighboring keratinocytes. This breaking down process is just as complex as the structure itself, and involves interactions between MMPs, integrins, growth factors, and structural proteins. In the normal resting state, laminin-5 is bound to α6β4 integrin, the latter linking the intracellular keratin filaments of keratinocytes to the basement membrane. In large part due to the interactions of integrins (including their phosphorylations status) with the ECM and receptor clustering on the surface of keratinocytes, there are important morphological changes (such as lamellipodia formation) that are required for keratinocyte locomotion.3,40–42 Molecular GTPases switches (Rho, Rac, Cdc42) are involved. Partially as a result of phosphorylation of the α6β4 integrin, α3β1 integrin promotes lamellipodia formation and keratinocyte locomotion. It has often been stated that migration of keratinocytes is key to resurfacing of the wound and that in fact such resurfacing is not dependent on proliferation.3 Overall, one must add, epithelial-mesenchymal transition (EMT), also important in cancer and epithelial adhesion and gain of mesenchymal features, empowers the epithalial cells to migrate in a way that is reminiscent of the embryonic stage. Recently, this process was reconstituted in vitro by exposure of epithelial cells to tumor necrosis factor-α (TNF-α), which led to the expression of vimentin, FSP1, and MMPs. The TNF-α mediated EMT may be secondary to bone morphogenetic proteins (BMP).43
In addition to the critical role of migration, keratinocytes do proliferate within the first hours after injury, and this is certainly more evident when the gap or size of the defect cannot be bridged temporarily with cell movement alone and is dependent on the cells beyond.2,3 By a week after injury, the repair process is well underway. Cytokines and growth factors (EGF, PDGF, TGF-β, FGFs, VEGF), matrix components, and MMPs continue to play an active role. In addition to the growth factors themselves, signaling proteins are critical. For example, certain kinases (MAPK) are activated in basal and suprabasal keratinocytes by further action of integrins or the release of interleukin (IL)-1α. Ion fluxes, including calcium levels and entry into the cells, are also critical to the overall process of keratinocyte migration and skin resurfacing.44 There is evidence that, in addition to lamellipodia extension, wound edge suprabasal keratinocytes may “leap-frog” over the basal cells near the wound. However, this system may not be very efficient, certainly not to the extent of the purse-string mechanism occurring in fetal healing and corneal healing, which is accompanied by other important differences in matrix deposition and growth factor profile.5,45 Wound contraction, rather than epithelialization, is common in major injuries, including burns. It is also the preferred mode of repair in many animal species, such as rodents. In experiments using knockout mice, MMP-13 has been implicated in keratinocyte migration, angiogenesis, and contraction, while the role of MMP-9 appears critical in keratinocyte migration.46
The remodeling of the ECM, as well as the movement of cells, is highly dependent on MMPs and serine proteases.47 A very important component of this dependence on MMPs is the MMPs-driven degradation of ECM and the resultant exposure of selective bioactive ECM segments that influence cell behavior, including migration and proliferation.47 The remodeling phase begins 5–7 days after injury.11 Upregulation of tPA and uPA are important for keratinocyte migration, which may depend on cross-talk and interactions between α3β1, keratinocytes, and collagen. These events lead to the induction of MMP-1 (collagenase-1 or interstitial collagenase), important in keratinocyte migration and epithelialization.11 MMP-9 plays a fundamental role in “cutting” Type IV and type VII collagen, which are essential components of the basement membrane and anchoring fibrils, and it also promotes inflammation and neutrophil migration. MMP-10 (stromelysin) breaks down other noncollagenous ECM components and facilitates migration.2,11,14,48 Other mediators, such as thymosin-β, have been found to upregulate MMPs during wound repair.49 Of critical importance during the remodeling process is the phenotypic switch in certain cell subpopulations from fibroblasts to myofibroblasts.2,50 Therefore, while the early process of healing relies heavily on matrix accumulation, which in turn facilitates cell migration, there is now a need to dampen the ECM formation and degrade it to a level that at least approximates the preinjury state. However, the remodeling phase is more than a breakdown of excess macromolecules formed during the proliferative phase of wound healing. Cells within the wound are returned to a stable phenotype, extracellular matrix material is altered (i.e., collagen type III to type I), and the granulation tissue that was so exuberant during the early phases of wound healing disappears.2,3
Over the last several years, considerable attention has turned to the role of stem cells from the follicular and interfollicular epidermis in wound healing. Using murine models, it has been reported that, at least after injury, cells from the hair-follicle bulge are recruited to the epidermis and migrate to the center of the wound.51 In subsequent studies, it has been shown that hair follicles form de novo after wounding and that epidermal cells in the wound assume a hair follicle stem cell phenotype. Some of these findings may be dependent on Wnt signaling.52
Extracellular Matrix (ECM)
In wound healing, the extracellular matrix (ECM) comprises four main components:47 (1) structural proteins (i.e., collagen, elastin); (2) multidomain adhesive glycoproteins (i.e., fibronectin, vitronectin, laminin); (3) matricellular proteins, including secreted protein acidic and rich in cysteine (SPARC), thrombospondin 1 and 2, tenascins, osteopontin; and (4) glycosaminoglycans (GAG), including hyaluronic acid and proteoglycans, that is, syndecans, perlecan (such as chondroitin sulfate and heparan sulfate). The glycosaminoglycan (GAG) hyaluronic acid (hyaluronan) is a particularly abundant component of the provisional matrix, and one whose deposition needs to be modified during the remodeling process. GAGs are often around other ECM proteins, including collagen and elastin.47 High levels of hyaluronic acid and fine reticular collagen are present in the early embryo, where they are thought to offer less resistance to cell migration.53 Indeed, embryonic wound repair is characterized by a hyaluronic acid–rich environment, thought to be at least in part responsible for the “scarless” healing of embryonic wounds. Fibroblasts from early granulation tissue produce large amounts of hyaluronic acid, and proliferating cells express CD44, which is the receptor for this GAG molecule.54,55 Besides the issue of offering less resistance to cell movement, hyaluronic acid may stimulate cell motility by altering cell-matrix adhesion. One example of this is that hyaluronic acid weakens the adhesion of heparan sulfate and fibronectin.56 Perhaps more importantly from a physical/spatial standpoint is that hyaluronic acid creates a highly hydrated structure leading to tissue swelling and interstitial spaces, and thus an environment more conducive to cell movement.2,57 The effects of hyaluronic acid are also regulated by growth factors and cytokines. Upregulation of the hyaluronic acid expression and its receptors (for example RHAAMM) by TGF-β1 stimulates fibroblast motility.58 Eventually, as remodeling occurs, hyaluronic acid is degraded by hyaluronidase and replaced by sulfated proteoglycans, which contribute a stronger structural role in late granulation tissue formation and in scars, while being less able to stimulate cellular movement. Two major proteoglycans, chondroitin-4-sulfate and dermatan sulfate, are produced by mature scar fibroblasts.
Three main classes of collagens are present normally in connective tissue: (1) fibrillar collagens (types I, III, and V), (2) basement membrane collagen (type IV), and (3) other interstitial collagens (types VI, VII, and VIII). These are examples of the different types of collagen present in skin. Importantly, however, the fibrillar collagens serve as the major structural collagens in all connective tissues.2,57,59 During the initial phases of wound repair, it appears that the wound tends to recapitulate the processes involved in embryogenesis.45 Thus, granulation tissue is initially comprised of large amounts of type III collagen, which is a minor component of adult dermis and indeed is present in larger amounts in fetal wound repair. During the phase of remodeling, type III collagen is gradually replaced by Type I collagen. Type I collagen replacement is associated with increased tensile strength of the scar. However, the final tensile strength of a scar is only about 70% of that of preinjured skin.3,57 The process of converting the collagen content of the dermis from type III to type I collagen is controlled by interactions involving synthesis of new collagen with breakdown of old collagen.14 Key to this process of conversion are matrix metalloproteinases (MMPs) and, specifically, the collagenases.
Matrix-degrading metalloproteinases (MMP) are proenzymes that need to be activated, and are considered to be the physiologic mediators of matrix degradation.48 The prototypic MMP is interstitial collagenase, but over 20 such enzymes (zinc-dependent endopeptidases) have been described.11 We have already mentioned these critical proteinases in the context of keratinocyte migration, but it is important to go over them in more detail. Five groups of these enzymes have been identified: (1) collagenases, (2) gelatinases, (3) stromelysins/matrilysins, (4) membrane-type MMPs, and (5) other types. Table 248-2 is a summary of certain MMPs that have a prominent effect in wound healing, and which supplements this discussion. The collagenases include interstitial collagenase (fibroblast collagenase, MMP-1), which acts on collagens I, II, III, VII, and X. Collagen type II is a particularly good substrate for MMP-1. Another important member of the collagenase class is neutrophil collagenase (MMP-1), which also degrades type II and III collagens but is particularly active against type I collagen. Gelatinases break down denatured collagen (gelatin). Among the most important gelatinases are gelatinase A (MMP-2), which breaks down gelatins, collagen IV, and elastin. Another key gelatinase is gelatinase B (MMP-9), which is produced by many cell types, including macrophases, neutrophils, and keratinocytes. Stromelysins have a relatively broad substrate specificity. Both stromelysin 1 (MMP-3) and 2 (MMP-10) act on proteoglycans, fibronectin, laminin, gelatins, and collagens III, IV, and IX. Another member of the stromelysin family, matrilysin (MMP-7) degrades mainly fibronectin, gelatins, and elastin. A member of the MMPs family is epilysin (MMP-28), which appears to be produced by proliferating keratinocytes distal to the wound edge. It may be needed to restructure the basement membrane.3,48,57 Deficiency of MMP-7 (matrilysin-1), which regulates inflammation, epithelialization, and inhibits apoptosis, is reported to lead to the most severe wound healing defect associated with MMPs.11
Effect | Common Names | Corresponding MMP Designation | Some Specific Effects |
|---|---|---|---|
Keratinocyte Proliferation and Migration | Collagenase 1 Gelatinase A Stromolysin 2 Matrilysin-1 Epilysin | MMP-1 MMP-2 MMP-10 MMP-7 MMP-28 | Increased migration |
Endothelial cell (EC) migration | Collagenase 3 Gelatinase A MT1-MMP | MMP-13 MMP-2 MMP-14 | Increases (EC) migration Needed for angiogenesis Needed for angiogenesis |
Cell migration | Stromolysin 1 Stromolysin 2 Matrilysin-2 | MMP-3 MMP-10 MMP-26 | Required for excisional wounds |
Inflammation | Collagenase 2 Gelatinase A Gelatinase B Matrilysin-1 | MMP-8 MMP-2 MMP-9 MMP-7 | Anti-inflammatory Anti-inflammatory Promotes inflammation |
Neutrophil Migration | Gelatinase B MT6-MMP | MMP-9 MMP-25 | Increases neutrophil migration |
Apoptosis | Collagenase 2 MT1-MMP MT2-MMP MT6-MMP | MMP-8 MMP-14 MMP-15 MMP-25 | Prevents apoptosis Antiapoptotic |
Moist Wound Healing and the Repair Process
One of the most important clinical observations of the last several decades is the realization that wounds kept moist re-epithelialize faster.60–63 The evidence for this is best for acute wounds. However, even in chronic wounds, the moisture-retentive dressings that have been developed to create moist wound conditions do lead to a number of desirable outcomes, such as pain control, painless autolytic debridement, and stimulation of granulation tissue.63–66 There have been fears that keeping the wound moist will cause infection, but this fear is unfounded.67–69 Nevertheless, occlusion of the wound is still contraindicated in the presence of infection. There are a variety of wound dressings available to the clinician and which fit the particular clinical situation.65,66,70,63 The main types of dressings include transparent films, hydrocolloids, foams, gels, alginates, and the relatively new collagen products (Table 248-3). In determining the most appropriate dressing for a particular wound, the clinician must take into consideration the need for absorption of excessive exudate (foams and alginates), whether the wound is too dry and needs additional moisture (gels or hydrogel materials) and whether the wound and its epithelial edges can tolerate the often subtle but serious trauma that comes from removal of adhesive dressings such as films. Thin contact layers, consisting of different polymeric materials with perforations, allow wound fluid to escape and are useful in preventing tissue injury upon dressing changes by minimizing removal of the primary dressing in direct contact with the wound.
Products and Properties | Other Advantages | Disadvantages | Other Indications |
|---|---|---|---|
Absorb exudate | |||
ALGINATES | Hemostatic, nonadherent, fewer dressing changes | Require secondary dressings; foul-smelling gel | Highly exudative wounds, partial- or full-thickness wounds, postoperative wounds |
FOAMS | Conforms to body contours, applicable to many wounds | Opaque, require secondary dresssings, may adhere to the wound | Partial-thickness exudative wounds, pressure relief. May be used in many uncertain clinical situations |
HYDROFIBERS | Soft, interacts with exudate to form gel | May be opaque and require secondary dressings, sometimes difficult to remove | Deep wounds, packing |








