Management of Nerve Injuries and Compressive Neuropathies of the Upper Extremity
Scott A. Mitchell
Kodi Azari
PERIPHERAL NERVE INJURY
The peripheral nerve is a hierarchical structure in which the axon is the basic subunit. The endonerium surrounds individual myelinated axons or groups of unmyelinated axons. Collections of axons are gathered into fascicles by a layer of perineurium. The fascicle is the smallest subunit of peripheral nerve that can be surgically dissected. Within most peripheral nerves, fascicles are grouped together by condensations of internal epineurium. For instance, as the ulnar nerve approaches the wrist, distinct fascicular groups bound for the dorsal sensory, deep motor, and superficial sensory branches may be identified. The outermost layer of connective tissue, the external epineurium, encases the fascicular groups to form the peripheral nerve. The epineurium is typically surrounded by loose areolar tissue permitting nerve excursion with joint motion.
Common etiologies of peripheral nerve injury include penetrating trauma, traction, compression, electrical, and thermal injuries. Many injuries reflect a combination of these mechanisms. A variety of secondary processes, including infection, ischemia, and fibrosis, may contribute to further damage. Although the peripheral nerve may be injured in a variety of manners, it has a limited arsenal of responses. There are two basic pathophysiologic responses to trauma—demyelination and axonal degeneration. This primary distinction forms the basis for most classifications of nerve injuries. Mild injuries result in local demyelination along otherwise intact axons. Repair of the myelin sheath by residing Schwann cells typically restores function. More severe injuries affect the axons themselves, resulting in degeneration of the axons distal to the site of injury through the process of Wallerian degeneration. Regeneration of injured axons and re-innervation of distal targets are required to restore function.
Classification of Nerve Injury
There are two commonly used classification schemes for peripheral nerve injuries (see chapter 36). Seddon divided peripheral nerve injuries into three subtypes: neurapraxia, axonotmesis, and neurotmesis.1 Neurapraxia refers to a conduction block in which axons remain in continuity. In its mildest form, temporary neural ischemia produces a reversible physiologic conduction block. More severe forms result from focal demyelination at the site of injury. Axons remain anatomically intact but are unable to conduct across the injured segment. Prognosis for spontaneous recovery is typically good, although with more significant injuries remyelination may require several weeks. In the absence of secondary injury, full functional recovery can be expected within 2 to 3 months.
Axonotmesis refers to the loss of continuity of the axons within a peripheral nerve; however, all layers of the connective tissue architecture, including the endoneurial tubes, remain intact. Wallerian degeneration, a programmed involution of axonal segments, occurs distal to the site of injury. This process clears the endoneurial tubes of axonal and myelin debris in preparation for axonal regeneration. Axonal regrowth begins near the site of injury and progresses under ideal conditions at a rate of 1 mm/day. As the endoneurial tubes remain intact to guide regeneration, prognosis for recovery is favorable and functional recovery is anticipated.
Neurotmesis implies complete nerve transection with disruption of axons and all layers of investing connective tissue. There is degeneration of all axons distal to the site of injury as well as a physical separation of the nerve ends. Scar tissue formation within the intervening gap blocks advancement of the regenerating axons and results in a neuroma. Recovery will not occur unless the transected stumps are surgically coapted.
Sunderland expanded Seddon’s classification to delineate five degrees of nerve injury.2 First- and second-degree injuries are analogous to Seddon’s neurapraxia and axonotmesis, respectively. With third-degree injury, there is loss of continuity of the axons as well as the endoneurial tubes. The fascicular architecture and investing perineurium remain intact. Axonal regeneration will occur; however, specificity of reinnervation is compromised due to endoneurial disruption and prognosis is less favorable than with second-degree injuries. Fourth-degree injury refers to loss of continuity of the axons, endoneurial tubes, and perineurium, with only the outer epineurium remaining intact. This degree of injury often produces a neuroma in continuity due to intra-neural scaring that precludes significant spontaneous recovery. Fifth-degree injury, analogous to Seddon’s neurotmesis, refers to complete nerve transection.
During the early clinical evaluation of patients with traumatic peripheral nerve injuries, it is not possible to distinguish among these subtypes. The mechanism of injury may provide some guidance. Penetrating trauma tends to produce neurotmesis injuries. The zone of injury is typically small with sharp lacerations but may be more extensive with dull lacerations as stretching and tearing of tissues become more prominent. Traction tends to produce axonotmesis with a relatively extensive zone of injury. Acute compression generally produces neurapraxia due to local contusion. The surrounding inflammatory response, however, can contribute to subsequent scarring, chronic compression, and eventual axonal injury.
Electrodiagnostic Studies.
Electrodiagnostic studies are useful objective tests to classify nerve injuries and identify early stages of recovery. Testing consists of two components: nerve conduction studies (NCSs) and electromyography (EMG). NCSs measure signal transmission along large myelinated axons, whereas EMG measures spontaneous and induced electrical activity within target muscles.
With neurapraxic or demyelinating lesions, NCSs demonstrate a conduction block across the zone of injury. With complete injuries, when the nerve is stimulated proximal to the lesion, distal potentials are absent. With incomplete injuries, a characteristic feature of demyelination is slowing of
conduction across the lesion. When stimulated below the site of injury, however, conduction along the distal nerve segments remains normal. EMG demonstrates decreased voluntary motor unit action potentials. A defining feature of neurapraxic lesions on EMG is that no fibrillations or denervation changes develop regardless of the time since injury.
conduction across the lesion. When stimulated below the site of injury, however, conduction along the distal nerve segments remains normal. EMG demonstrates decreased voluntary motor unit action potentials. A defining feature of neurapraxic lesions on EMG is that no fibrillations or denervation changes develop regardless of the time since injury.
With axonal injury, including axonotmesis and neurotmesis, conduction both above and below the site of injury is disrupted due to axonal damage. Conduction abnormalities in the distal segment are not present acutely but develop over the first 1 to 2 weeks as axonal degeneration ensues. A characteristic feature of axonal damage on NCS in cases of incomplete lesions is decreased amplitude of distal potentials with normal conduction velocity. EMG demonstrates denervation potentials and fibrillations in affected muscles 3 to 4 weeks following injury. Their presence identifies axonal injury but does not discriminate between axonotmesis and neurotmesis lesions.
Clinical Management.
Initial treatment decisions are based primarily upon injury mechanism and presumptive classification of the nerve lesion. Neurologic deficits in the context of penetrating trauma, even if incomplete, are assumed to represent neurotmesis injuries. Early surgical exploration is recommended. Primary repair is ideally performed within 72 hours of injury. For sharp, clean lacerations with minimal contusion of the nerve ends, primary repair may be performed at the time of initial exploration. If significant crush or avulsion is identified or in cases with unstable wounds, the nerve ends should be grossly approximated to prevent retraction. Definitive reconstruction should be delayed for 2 to 3 weeks to allow demarcation of the extent of nerve injury. Nerve grafts or conduits are typically required to span the resultant nerve gap.
For closed injuries in which nerve continuity is uncertain, an initial period of observation is recommended. Treatment decisions are based on the assumption that neurapraxic and axonotmetic lesions will recover spontaneously and these lesions are best treated nonoperatively. As early as 1 to 2 weeks following injury, electrodiagnostic studies can distinguish neurapraxic from axonal injuries. With neurapraxia, conduction distal to the site of injury remains intact, whereas with axonal injury, distal conduction is impaired due to axonal degeneration. Early studies may also determine whether a lesion is complete or incomplete. The presence of even a small number of motor units under voluntary control defines an incomplete lesion. This indicates that the nerve has not been completely transected and that surgery is less likely to be required.
Electrodiagnostic studies are traditionally repeated 4 to 6 weeks following traumatic injury. EMG evidence of target muscle denervation identifies the presence of axonal injury. However, the only way to differentiate axonotmesis from neurotmesis injuries, aside from surgical exploration, is to monitor the patient for signs of recovery. This critical distinction relies on the return of motor unit potentials following axonal regeneration, confirming the presence of an axonotmesis lesion. However, this is a retrospective diagnosis and intervention cannot be delayed indefinitely. There are a number of temporal factors to consider in determining the optimal timing of intervention for non-recovering deficits. With neurapraxic injuries, remyelination may require up to 8 to 12 weeks. With axonotmesis injuries, axonal regeneration proceeds at a rate of 1 mm/day under ideal conditions. Depending on the site of injury and distance to the nearest target muscle, evidence of early re-innervation will often become apparent within 3 to 6 months. However, a competing process occurs during this time that must also be considered. Irreversible atrophy gradually occurs in denervated muscle (estimated rate of loss of 1% per week) such that significant motor recovery is unlikely beyond 12 to 18 months. For this reason, if there is no clinical or electrodiagnostic recovery evident by 3 to 6 months after injury, surgical exploration is generally recommended. For chronic nerve injuries presenting beyond the 12- to 18-month window, tendon transfers should be considered.
Two common clinical scenarios deserve special mention. Gunshot wounds are considered apart from other penetrating traumas. Most deficits are secondary to concussive effects rather than direct laceration. The potential for spontaneous recovery is favorable and a period of observation as with closed injuries is recommended. Nerve deficits occurring in association with fractures are managed primarily based on the fracture. With closed fractures, 70% to 80% of nerve deficits are neuropraxic and may be managed nonoperatively. However, if internal fixation is indicated, nerve exploration may be reasonably performed during fracture exposure. The incidence of nerve laceration increases with open fractures. Early nerve exploration at the time of wound debridement is recommended. Definitive reconstruction should await fracture repair and stable wound coverage.
COMPRESSIVE NEUROPATHIES OF THE UPPER LIMB
Compressive neuropathies are among the most common clinical problems encountered in the upper limb. Each of the major peripheral nerves demonstrates a predilection for entrapment in specific anatomic regions. Although presentations vary based on the affected nerve and site of entrapment, the common pathophysiologic pathway reflects the mechanical and ischemic effects of chronic compression. In their early phases, compressive neuropathies resemble focal neurapraxias. With short-term compressive pressures of 30 mm Hg, there is disruption of intra-neural blood flow and loss of slow axonal transport.3 This manifests clinically as temporary paresthesias and muscle weakness. Increased vessel permeability following an episode of compression also causes intra-neural edema, which leaves the nerve more susceptible to further insults.3 More prolonged compression affects the myelin sheath, leading to focal demyelination beginning with large diameter sensory and motor fibers. In their chronic forms, compressive neuropathies are characterized by a mixture of demyelination and axonal loss. It is thought that the inflammatory reaction to compressive episodes, in conjunction with chronic ischemia and impaired axonal transport, ultimately lead to progressive fibrosis and axonal damage. The critical pressure or duration of compression required to produce axonal injury remains unknown.
COMPRESSIVE NEUROPATHIES OF THE MEDIAN NERVE
Carpal Tunnel Syndrome
The carpal tunnel refers to the fibro-osseous canal bounded by the concave bony arch of the carpus and the transverse carpal ligament (TCL). Although the tunnel is open-ended both proximally and distally, it behaves much like a closed compartment physiologically. Contents of the carpal canal include the median nerve and tendons of the flexor digitorum superficialis (FDS) and flexor digitorum profundus (FDP), and the flexor pollicis longus (FPL). The median nerve typically runs in a superficial and radial position (Figure 74.1). Near the distal aspect of the carpal tunnel, the median nerve divides into sensory branches to the thumb, index, middle, and radial ring fingers and the recurrent motor branch to the thenar musculature. Anatomic variations in the course of the recurrent motor branch are classified as extraligamentous, subligamentous, and transligamentous patterns, the latter being most vulnerable to injury during carpal tunnel release. The palmar cutaneous nerve arises 5 cm proximal to the wrist crease. It passes superficial to the TCL to supply the skin over the thenar eminence.
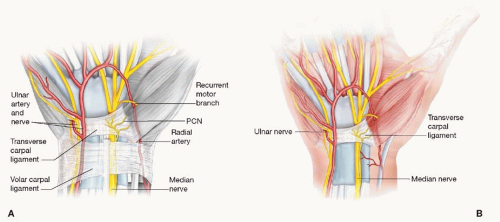 FIGURE 74.1. The median nerve in the carpal tunnel. (A) Note the superficial and radial position of the median nerve beneath the transverse carpal ligament. (B) The nearby ulnar neurovascular bundle passes superficial to the transverse carpal ligament as it courses through Guyon’s canal. |
Carpal tunnel syndrome (CTS) represents the most common compressive neuropathy encountered clinically. Common comorbid conditions associated with increased risk of developing CTS include advanced age, female gender, obesity, diabetes, and pregnancy. A variety of additional etiologic conditions have been implicated, including hypothyroidism, rheumatologic and autoimmune diseases, alcoholism, and renal failure. Compression may also result from space-occupying lesions within the canal such as proliferative tenosynovitis, hematoma, tumors, or ganglion cysts. Displaced distal radius or carpal injuries may also diminish canal volume. Although CTS is often viewed as an occupational disorder, a causative relationship underlying the commonly cited association with cumulative or repetitive work activity such as keyboarding has not been objectively demonstrated.
The history and physical examination are cornerstones for diagnosis. Patients classically report intermittent numbness and paresthesias in the radial digits. Symptoms may be exacerbated with activities involving prolonged wrist flexion or extension and are characteristically relieved by shaking the hand. Nocturnal symptoms are considered a hallmark of CTS, and their absence should invite suspicion for alternate causes. In more severe cases, numbness and/or paresthesias become constant. Frequent dropping of objects and loss of coordination in the hand may be reported and likely reflect the combination of thenar weakness and impaired sensibility in the radial digits.
Examination may demonstrate decreased light touch sensation in the median innervated digits. Objective threshold tests including Semmes-Weinstein monofilament testing are more sensitive to detect early sensory loss compared with innervation density tests such as two-point discrimination. With advanced disease, weakness and atrophy of the thenar musculature develop. Several examination maneuvers have been described to aid in the diagnosis of CTS. Tinel’s nerve percussion test, Phalen’s wrist flexion test, and Durkan’s nerve compression test are among the most common tests. Provocative tests should reproduce paresthesias in the median nerve distribution.
Electrodiagnostic studies remain the primary objective test to diagnose CTS and are considered by many to represent the reference standard. The hallmark findings of compressive neuropathies on NCSs are an increase in distal latency and a decrease in conduction velocity. Although standards vary, distal motor latencies of greater than 4.5 ms and/or distal sensory latencies of more than 3.5 ms are generally considered diagnostic. A decrease in amplitude of distal potentials, indicative of axonal loss, may be seen in more severe cases. EMG may also demonstrate increased insertional activity, fibrillations, and denervation potentials in the thenar musculature with advanced disease.
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


