Fig. 21.1
Enlargement of the fingers or hand secondary to vascular malformations: (a) A lymphatic malformation of the tip of the middle finger. (b) An arteriovenous malformation of a finger leading to macrodactyly. (c) A venous malformation causing macrodactyly of the middle finger. (d) Muscle hypertrophy of the hand associated with a lymphatic malformation
The relative growth of the digit, compared to unaffected digits, may be considered as either (1) static or (2) progressive [2, 21], and may be symmetric or asymmetric [18, 22] (Fig. 21.2).
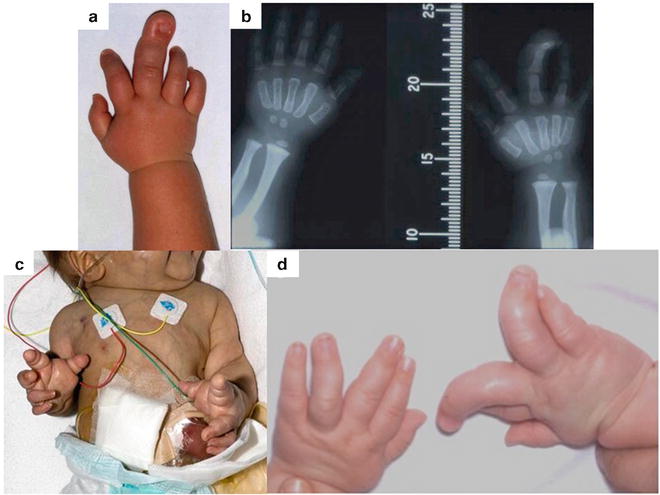
Fig. 21.2
The multiple variations of upper limb macrodactyly: (a) Progressive unilateral macrodactyly of the right middle finger. (b) Radiograph of the hands with the superimposition of a measure to allow for serial growth recording. (c) Bilateral, symmetrical macrodactyly in a child with an “unknown” syndrome which was fatal. (d) Bilateral, asymmetric macrodactyly
(a)
In the static type, enlargement is present at birth and the affected limb grows in proportion to the child.
(b)
In the progressive type, some overgrowth may be noted at birth, but at around 2 years of age there is evidence of slow, unrestricted, and disproportionate digital enlargement, which continues until closure of the epiphyses [23].
In our review of 32 patients with macrodactyly, approximately two-thirds had the static type, which is different from the findings of other authors [3, 4]. In our cohort of 20 patients, 6 required no surgical intervention. It has been observed that such patients usually present later with good function [21] but we did not note any significant difference in age at presentation between the static and progressive subtypes, although it was noted that those with static disease required fewer operations overall, which was significant. These findings are echoed by Cerrato et al. [4]. It is difficult to classify into static or progressive type before the age of two, as the only significant indicator of the prognosis in macrodactyly is by regular observation in the first few years of life.
The most comprehensive classifications to date are based upon the original work of Kelikian, and later modified by Dell, Flatt, and Upton [7, 23–26] (Table 21.1). Lipomatous macrodactyly is the most common form of overgrowth in the literature and is differentiated from NTOM by the absence of infiltration of the digital nerves upon microdissection and neurovascular structures are of normal caliber. NTOM was introduced by Kelikian [26] to differentiate the digital nerve involvement from that observed in neurofibromatosis-associated macrodactyly, and to emphasize the relationship between the enlargement of the nerve along with the bone and soft tissues. It is a common type of macrodactyly that is unilateral in 90 % of cases. As with lipomatous macrodactyly, it does not usually show a pattern of inheritance, nor association with other malformations [25–27]. The median nerve territory is more often affected, with 85 % of cases of macrodactyly affecting the thumb, index, or middle fingers [27]. Our recent review shows a similar distribution with involvement of these digits in 63 % of cases and out of the 32 cases presented, 27 were lipomatous or NTOM macrodactyly [3]. Cerrato et al. presented 21 cases of macrodactyly, of which 12 were NTOM and 9 lipomatous. No significant difference in patients, progression, number of operations, distribution, or associated anomalies was found [4].
Table 21.1
Chronologic development of the different classification systems of macrodactyly
Author(s) (year of publication): | Holmes (1869) | Richardière (1891) | De Laurenzi (1962), Barsky (1967) | Kelikian (1974) | Upton (1990), Flatt (1994) | Upton (2006) |
|---|---|---|---|---|---|---|
Classifications of macrodactyly | Symmetric | True | Static | Lipomatous macrodystrophy | Gigantism and (nerve oriented) lipofibromatosis | Nerve territory oriented macrodactyly |
Asymmetric | False | Progressive | Neurofibromatosis | Gigantism and neurofibromatosis | Lipomatous macrodactyly | |
Nerve territory orientated macrodactyly | Gigantism and digital hyperostosis | Neurofibromatosis | ||||
Hyperostotic variety | Gigantism and hemihypertrophy | Hyperostosis | ||||
Hemihypertrophy | ||||||
Proteus syndrome | ||||||
Vascular malformations |
Digital hyperostosis, or hyperostotic digital gigantism, also described by Kelikian, is a rare form of macrodactyly that is non-hereditary, and may present later [26, 28]. There is bilateral enlargement of the digits, which may be symmetric or asymmetric, without gross enlargement of the digital nerves or fat, but it can present in the median nerve distribution, with concurrent NTOM [7, 29]. Palpable periarticular osteochondral masses arise from the volar plates of the metacarpals and phalanges, similar to the pattern seen in neurofibromatosis, and can lead to profound loss of motion [7, 23]. The joint involvement seen in hyperostosis and neurofibromatosis or Proteus syndrome will presumably lead to poorer functional outcomes, although this is not evidenced in the literature. The remaining subclasses associated with other anomalies or syndromes are discussed later.
The real value of a medical classification is to provide prognostic information, or to group patients for prospective analysis. With a confused variety of systems available for macrodactyly built upon phenotypic, intra-operative, histological, radiological, or genetic findings, the most effective classification should be based upon outcomes. It has been shown that patients with static disease need significantly fewer operations than those with progressive, although it must be acknowledged that the patient cohorts on which these assumptions are made were small and prone to variability [3, 4]. Macrodactyly associated with other anomalies or syndromes may also have poor functional outcomes due to joint involvement, flexion contracture, and other morbidity secondary to the syndrome involved.
With this established, we suggest a new three-layer classification based upon (1) growth progression, (2) associated anomalies, and (3) structures involved (Table 21.2). This encompasses all types of macrodactyly reported to date and can provide information about prognosis and allow grouping of similar patients for subsequent analysis. As such, a (1) static–(2) isolated–(3) lipomatous macrodactyly can be assumed to have a better prognosis in terms of function, fewer operations, and fewer surgical complications than a (1) progressive–(2) syndrome–associated–(3) hyperostotic macrodactyly.
Table 21.2
A proposed new system for the classification of macrodactyly
Characteristic | Classification | |||
|---|---|---|---|---|
1. Growth | Static | Progressive | ||
2. Associations | Isolated | Associated syndrome or anomalies | ||
3. Structure | Lipomatous | Nerve territory orientated | Hyperostotic | Vascular malformation |
Incidence
Macrodactyly is classed as a rare disease by the Office of Rare Disease Research, and thus affects less than 200,000 people in the USA [30]. With rare conditions, such as macrodactyly, a true population incidence is hard to calculate. The classic description by Flatt of an incidence of 0.9 % of all congenital hand anomalies is based upon the author’s personal study of 2,758 patients, with 28 cases of macrodactyly in 26 patients [7]. This figure, although widely published, has no reference to the overall incidence in the general population. A similar number was found in Hong Kong in the 1980s, with two cases of macrodactyly from a cohort of 326 patients with congenital upper limb anomalies, equating to 0.5 % [31].
Congenital anomalies occur in 1–2 % of newborns, with 10 % of these affecting the upper limb [7]. National population studies from Sweden have also shown an incidence of upper limb anomalies to be approximately 1/500 [32–34] and one can therefore extrapolate the incidence of upper limb macrodactyly (~1 % of the total) to be around 1/50,000, but this is based upon many assumptions. Lower limb macrodactyly has been estimated to have an incidence of 1/18,000 [35]. In a large UK teaching hospital, one expects to see one to two new cases of upper limb macrodactyly per year, which echoes historic findings [36].
Associations
There are many reported associations of macrodactyly with other clinical signs or syndromes. In isolated non-syndromic macrodactyly, there can be concurrence of local anomalies such as syndactyly (occurring in 10 % of patients) [29] and clinodactyly or curvature of the enlarged digit. There is also a very rare entity of syndactyly associated with dorsal macrodactyly (Fig. 21.3)
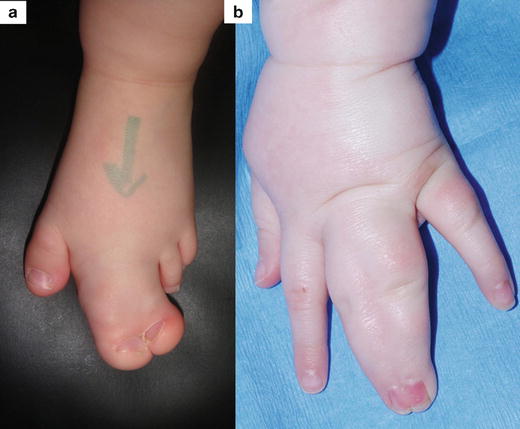
Fig. 21.3
Non-syndromic macrodactyly with associated malformations: (a) Macrosyndactyly of the second and third toes. (b) Subtle dorsal macrosyndactyly
Of the many reported syndromes that have presented with macrodactyly, none have it per se as a syndrome-defining feature. It is usually classified as part of an overgrowth component of the disease, which can be classified into (1) phakomatoses, (2) osteochondrodysplasias, (3) specific overgrowth syndromes, or (4) secondary to a vascular anomaly (Table 21.3).
Table 21.3
A classification for syndromes that have been associated with macrodactyly
Syndromes associated with macrodactyly | |||
|---|---|---|---|
1. | Phakomatoses | Neurofibromatosis type 1 and 2 | |
Hamartoma syndromes | Proteus syndrome | ||
Tuberous sclerosis | |||
2. | Osteochondrodysplasias | Enchondromatoses | Maffucci syndrome |
Ollier syndrome | |||
Monostotic fibrous dysplasia | |||
3. | Specific overgrowth syndromes | Beckwith–Wiedemann syndrome | |
4. | Vascular anomalies | Vascular malformations | Klippel–Trénaunay syndrome |
Maffucci syndrome | |||
CLOVES syndrome | |||
1.
The phakomatoses (or “neurocutaneous syndromes”) include (a) neurofibromatosis type 1 and 2 (NF1 and NF2) and (b) the hamartoma syndromes.
(a)
NF1 and NF2 have both been associated with macrodactyly [7, 23, 25]. NF1 is the most commonly reported syndrome presenting with digital enlargement, as well as enlargement involving the upper and lower limbs, torso, and head and neck. It has an autosomal dominant inheritance pattern, with an incidence of 1/3,000 [37–39] and has specific diagnostic criteria [40]. Digital enlargement is frequently bilateral and presents in a similar manner to the previously described NTOM, or can be related to plexiform schwannomatosis [41]. It may also present with features of hyperostosis [25]. As has been shown in hyperostotic macrodactyly, bony involvement can lead to limitation of movement and function. Growth is usually progressive and resection of the neurofibromas, or involved nerves, has been shown to limit advancement of the disease [42–44]. One must always consider the risk of malignant transformation and development of neurofibrosarcoma in the peripheral nerve [45, 46].
NF2 also has an autosomal dominant inheritance pattern but is 20 times less common than NF1. It manifests as bilateral vestibular schwannomas, spinal cord meningiomas or ependymomas and cataracts [47, 48]. Peripheral nerve involvement is rare, but macrodactyly secondary to a NF2 peripheral nerve schwannoma has been reported [49].
(b)
Hamartomas are benign focal malformations that resemble a neoplasm in the tissue of its origin [50]. Multiple hamartomas are associated with syndromes such as Proteus syndrome and tuberous sclerosis (Fig. 21.4), both presenting with soft tissue and bone overgrowth [51, 52]. Proteus syndrome was described as a discrete clinical entity in 1979 [53] and assigned its name in 1983 [54] with reference to the Greek god who was gifted with the power to change his appearance at will. It is rare, and the first description is now attributed to Treves with his presentation of Joseph Merrick (the Elephant Man) to the Pathological Society of London in 1885 [55, 56]. It is not inherited, and displays genetic mosaicism [57–60]. After neurofibromatosis, it is the most widely reported syndrome associated with macrodactyly [61–65]. In this condition, macrodactyly can be highly variable, progressive, and asymmetric. Disproportionate growth throughout the body begins between the ages of 6–18 months and leads to severe overgrowth and flexion contractures and the hands, which, when combined with glabrous hyperplasia can preclude functional use of the hand [23, 66].
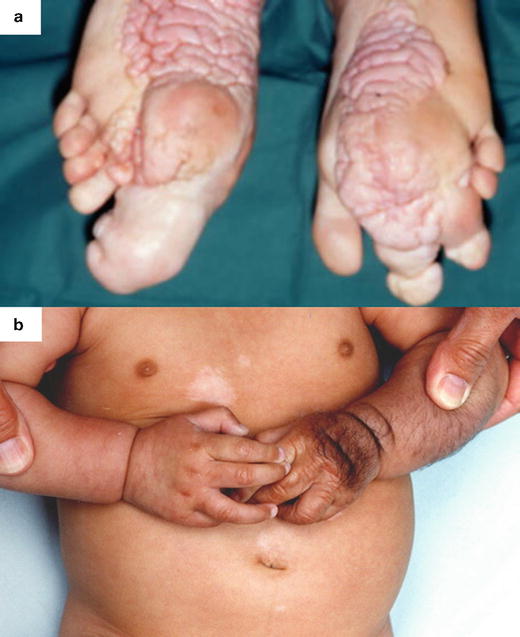
Fig. 21.4
Macrodactyly in association with hamartoma syndromes: (a) Bilateral macrodactyly of the toes and thickening of the plantar surfaces of the feet associated with Proteus syndrome. (b) Enlargement of the left hand associated with tuberous sclerosis
Tuberous sclerosis complex is an autosomal dominant disorder characterized by hamartomatous malformations in various organs such as brain, kidney, heart, and lung [67, 68]. Macrodactyly is rarely reported in association, with only ten cases identified up to 2000 [69]. There is a hyperostosis with cortical bone cysts, although the joints appear to be spared [70, 71]. The patients may have associated symptoms such as epilepsy or learning disability as a result of the primary diagnosis of tuberous sclerosis [72, 73].
2.
The osteochondrodyplasias that may present in infancy include (a) the enchondromatoses and (b) fibrous dysplasia, which may present with digital enlargement secondary to bone overgrowth.
(a)
Muffuci syndrome and Ollier disease are both enchondromatoses, characterized by multiple enchondromas that are almost exclusively localized in the metaphysis of long bones and in the small bones of the hands and feet [74–77]. Enchondromas can result in severe growth abnormalities (more severe than those observed in multiple exostosis) and fingers often show irregular morphology and size, although are rarely reported in the literature [76, 78]. Radiological findings include ovoid, cystic, and highly radiolucent lesions, elongated parallel to the major axis of the bone, originating near the physis and migrating towards the diaphyses with growth [76, 79, 80]. Debulking of the enchondromas and hemangiomas forego amputation and can result in a hand that is improved in appearance and less prone to trauma [81].
(b)
Fibrous dysplasia may be monostotic or poylostotic in presentation, or have associated endocrinopathy in McCune–Albright syndrome. Rarely reported monostotic involvement in the digit [82] must be considered in the differential diagnosis of macrodactyly.
3.
Overgrowth syndromes can be associated with hemihypertophy, of which macrodactyly can be a component. Beckwith–Wiedemann syndrome (BWS) is a rare and complex disorder of overgrowth with undetermined inheritance [83]. It was classically described as macrosomia, macroglossia, and an abdominal wall defect [84–86], but more recently has been noted to include hemihyperplasia [87, 88]. Hemihypertrophy macrodactyly presents similarly to Proteus syndrome but with more uniform soft tissue overgrowth and muscular hypertrophy. The palm and hand are less enlarged in proportion to the ipsilateral forearm, due to increased muscle bulk, but in extreme cases can be fixed in flexion at the wrist with digits in ulnar deviation due to muscular imbalance, which becomes more obvious during adolescence [23]. Isolated muscle hypertrophy of the hand including muscular hyperplasia, aberrant muscles, ulnar drift of the fingers in the metacarpophalangeal (MP) joints, flexion contractures of the MP joints, and enlargement of the metacarpal spaces is extremely rare but we have seen two cases associated with a lymphatic malformation (see Fig. 21.1d) and localized gigantism of the upper limb (Fig. 21.5).
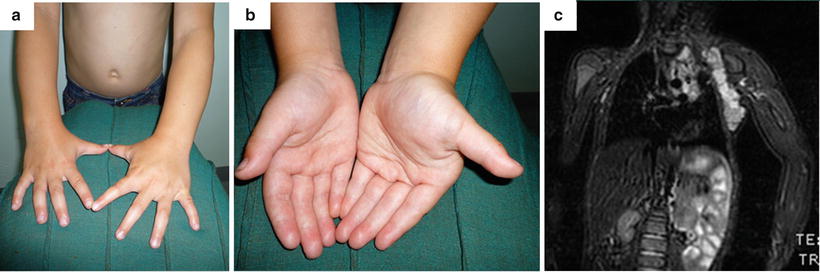
Fig. 21.5
Enlargement of the right hand musculature with sparing of the digits, in association with hemihypertrophy. (a) Dorsal view. (b) Anterior view. (c) Magnetic resonance imaging of the left upper limb showing generalized soft tissue hypertrophy
4.
Vascular anomaly syndromes that are associated with digital enlargement include Klippel–Trénaunay syndrome (KTS) and CLOVES syndrome. The anomalies that are present are vascular malformations (VM) as per the International Society for the Study of Vascular Anomalies [89, 90]. Macrodactyly in such cases may be diagnosed and managed with a different strategy, focusing on destruction or disruption of the VM prior to surgical debulking. KTS was described in 1900 with three characteristic features: a vascular nevus; hypertrophy of all of the tissues, particularly the skeleton; and ipsilateral varicosities [91, 92]. McGrory reviewed 108 patients with KTS and found 26 had macrodactyly (79 digits) of the upper or lower limb, amongst other congenital hand and foot anomalies. There was predilection for the radial side of the hand and medial foot [93].
CLOVES syndrome consists of congenital lipomatous overgrowth, vascular malformations, epidermal nevi, and skeletal abnormalities [94]. It can span the classifications, being part of either the overgrowth or vascular anomaly subset. The presence of high flow lesions in these patients suggests that it may be clinically related to KTS, and as such we have classified it here. The presence of truncal lipomatous mass and a characteristic pattern of macrodactyly differentiates CLOVES from other syndromic forms of overgrowth [94, 95]. The macrodactyly consists of progressive soft tissue overgrowth in predominance to bone overgrowth, which may be nonprogressive and non-distorting in nature [96, 97], which is markedly different from that found in Proteus syndrome. Review of the historical literature reveals probable CLOVES syndrome that may previously have been described as gigantism [95] or Proteus syndrome [94].
Genetics
The majority of isolated, non-syndromic macrodactyly, whether it of the progressive or static subtype, is sporadic in nature with no known underlying genetic causation recorded on the Online Mendelian Inheritance in Man database [98]. At present, there is no modern molecular insight into macrodactyly and there are no cellular or animal models of macrodactyly [99]. Candidate genes have been proposed (Table 21.4) which include those coding for bone morphogenetic proteins 5 and 7 (BMP5 & 7), transforming growth factor beta 3 (TGF-B3), Wnt (wingless-type) signaling pathway proteins (Wnt-2, Wnt-5A), pleiotrophin (PTN) [99], natriuretic peptide receptor 2 (NPR2) [100], and phosphoinositide-3-kinase (PI3K) [101].
Table 21.4
Genes associated with non-syndromic and syndromic macrodactyly
Gene | Chromosome location | Encoded protein | Syndrome | Gene(s) | Chromosome location | Encoded protein(s) | |
|---|---|---|---|---|---|---|---|
Non-syndromic macrodactyly | BMP5 | 6p12.1 | Bone morphogenetic protein 5 | Neurofibromatosis type 1 | NF1 | 17q11.2 | Neurofibromin |
BMP7 | 20q13.31 | Bone morphogenetic protein 7 | Neurofibromatosis type 2 | NF2 | 22q12.2 | Merlin | |
TGFB3 | 14q24.3 | Transforming growth factor beta 3 | Proteus syndrome | AKT1 | 14q32.33 | RAC alpha serine/threonine-protein kinase | |
WNT2 | 7q31.2 | Protein Wnt-2 | Proteus-like syndrome | PTEN | 10q23.31 | Phosphatase and tensin homolog | |
WNT5a | 3p14.3 | Protein Wnt-5a | SOLAMEN syndrome | PTEN | 10q23.31 | Phosphatase and tensin homolog | |
PTN | 7q33 | Pleiotrophin | Tuberous sclerosis | TSC1 | 9q34.13 | Hamartin | |
TSC2 | 16p13.3 | Tuberin | |||||
NPR2 | 9p13.3 | Natriuretic peptide receptor 2 | Beckwith–Wiedeman syndrome | CDKN1C, H19, IGF2, KCNQ1OT1 | 11p15.5 | Cyclin-dependent kinase inhibitor 1C | |
Insulin-like growth factor 2 | |||||||
PIK3CA | 3q26.32 | Phosphoinositide-3-kinase | CLOVES syndrome | PIK3CA | 3q26.32 | Phosphoinositide-3-kinase |
BMP5 and 7, TGF-B3, Wnt-2, and Wnt-5A are all overexpressed in macrodactyly, but PTN had the greatest fold-change when reported [99]. PTN is a promising candidate for the pathogenesis of macrodactyly because it promotes growth of nearly all the tissues affected by macrodactyly: PTN is necessary for neurite outgrowth and maturation in the central nervous system [102–104]. In the peripheral nervous system it promotes nerve regeneration following injury [105]. PTN is highly expressed in bone and cartilage, and is upregulated in response to mechanical loading [106–108], is an angiogenic factor, and supports endothelial cell proliferation [109].
Overproduction of C-type natriuretic peptide (CNP) due to a chromosomal translocation was reported to cause skeletal dysplasia associated with tall stature [110, 111]. In addition, acromesomelic dysplasia, characterized by dwarfism and short limbs, is caused by loss of function mutations in the NPR2 gene [112]. In a study by Miura et al., a three-generation family of tall stature and macrodactyly of the great toes had a gain of function mutation in the NPR2 gene [100].
PI3K is an upstream regulator of the AKT-mTOR cell-signaling pathway, which has been implicated in non-syndromic macrodactyly and CLOVES syndrome [101, 113]. The PI3K/AKT/mTOR pathway is important in apoptosis and carcinogenesis [114, 115] and muscular hypertrophy [116].
In syndrome-associated macrodactyly, especially those with an autosomal dominant inheritance pattern, individual genes have been identified (see Table 21.4). A mutation in the NF1 gene that encodes for neurofibromin leads to the development of NF1 [117, 118]. The NF2 gene encodes for merlin (also known as schwannomin) and mutations lead to the development of NF2 [47, 48, 119]. Although its exact function is unknown, merlin is likely also involved in controlling cell movement, cell shape, and communication between cells [120–122], with mutations leading to the development of schwannomas, which may be associated with macrodactyly [49].
Proteus syndrome is caused by a mutation in the AKT1 gene that encodes for RAC-alpha serine/threonine-protein kinase (AKT1), which regulates cell growth, proliferation, and apoptosis [123]. A mutation in AKT1 leads to the abnormal growth characteristics of Proteus syndrome [66, 124]. Mutations in the PTEN gene, which encodes for phosphatase and tensin homolog (PTEN), have been associated with asymmetric overgrowth but do not meet the strict guidelines for a diagnosis of Proteus syndrome [125–127]. Instead, these individuals have Proteus-like syndrome, which is considered part of a larger group of disorders called PTEN hamartoma tumor syndromes. Segmental overgrowth, lipomatosis, arteriovenous malformation, and epidermal nevus (SOLAMEN) syndrome, another variant within this group with mosaic PTEN mutations, has presented with macrodactyly [128].
Tuberous sclerosis complex is caused by mutations in the TSC1 or TSC2 genes, which encode hamartin and tuberin, respectively [67]. The proteins act as tumor suppressors, and with loss of function mutations leads to the growth of tumors in many different organs and tissues [68, 72], which may lead to macrodactyly [69]. The genetic causes of BWS are complex [83, 129], and involve several genes that are intrinsic to normal growth, including the CDKN1C, H19, IGF2, and KCNQ1OT1 genes [129–133]. The CLOVES syndrome is linked to PIK3CA gene mutations [113, 134] and is negative for PTEN gene mutations [94], which allows differentiation from similarly presenting PTEN hamartoma syndromes (e.g., SOLAMEN syndrome).
Imaging
Prenatal diagnosis of macrodactyly has been reported. Yuksel et al. present a case of macrodactyly of the second toe of the left foot, diagnosed at 24 weeks gestation on obstetric ultrasound scan (USS), with no other anomalies diagnosed [135]. Rypens et al. report a case of Proteus syndrome, diagnosed antenatally on USS, which presented with macrodactyly of the left middle finger and an associated massive axillary lymphangioma [136]. The evidence relating to the imaging of macrodactyly is not evident in the literature. In most cases, the diagnosis can be ascertained from the clinical history and examination after birth. If there is doubt as to the diagnosis, or if there is disproportionate growth of a previously unaffected digit, especially in adulthood, or after closure of the epiphyses, radiological investigation would be recommended.
Management
This section is inevitably anecdotal. Macrodactyly consists of a variety of conditions with many variations in presentation and growth patterns. It presents an almost unique situation whereby there is significant unpredictability of outcome with or without surgery. The management of macrodactyly demonstrates the need for very close observation of the child and detailed discussions with the parents and eventually the child throughout their growing years. It also requires considerable flexibility from a surgeon who has to consider the use of a wide variety of procedures and techniques. The aims of surgery are to allow a child to develop with minimum hindrance from their enlarged digit(s), e.g., by enabling them to be comfortable in shoes or to remove a digit, which is hindering their development of manipulative skills. This is done by attempting to reduce the disparity of the circumferential and longitudinal dimensions of the affected digit(s) when compared with the unaffected [25], with preservation of sensation, blood supply, and function, as far as is possible [23].
In terms of principles of surgery, the senior author currently uses a lateral approach to each individual digit tackling one side at a time with an interval of a few months between each operative procedure. With this approach, bone and soft tissues surgical reduction can be combined. Palmar or plantar soft tissue debulking can be carried out using a variety of incisions, including zigzag and longitudinal [7, 23, 26], as well as the use of skin grafts in the reconstruction [137]. The position of scars rarely causes a problem even when using a longitudinal plantar incision [3].
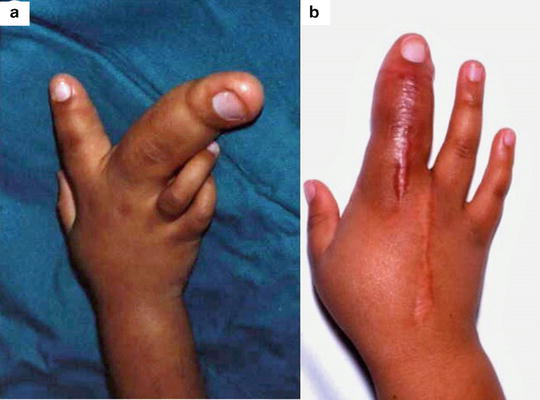
The need for repeated procedures must be highlighted in the cases of progressive macrodactyly. The eventual decision to carry out a ray amputation (Fig. 21.6) should not be considered a failure in management as it may take many years and several operations to arrive jointly at this decision, which can transform the quality of life for these patients. In the vast majority of cases of surgically treated macrodactyly, the functional and cosmetic outcome will be acceptable to the patient [3].
 Anesthesia Concerns in Congenital Anomalies of the Upper Extremity
General Skeletal Disorders
Anesthesia Concerns in Congenital Anomalies of the Upper Extremity
General Skeletal Disorders
 Therapy Management of Children with Congenital Anomalies of the Upper Extremity
Therapy Management of Children with Congenital Anomalies of the Upper Extremity
 Physical Medicine and Rehabilitation Management of Children with Congenital Anomalies of the Upper Extremity
Physical Medicine and Rehabilitation Management of Children with Congenital Anomalies of the Upper Extremity
 Ulnar Polydactyly and Ulnar Dimelia
Ulnar Polydactyly and Ulnar Dimelia
 Arthrogryposis
Arthrogryposis




Related posts:
Anesthesia Concerns in Congenital Anomalies of the Upper Extremity
General Skeletal Disorders
Therapy Management of Children with Congenital Anomalies of the Upper Extremity
Physical Medicine and Rehabilitation Management of Children with Congenital Anomalies of the Upper Extremity
Ulnar Polydactyly and Ulnar Dimelia
Arthrogryposis
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
